GI2019
Genome Informatics conference 2019
Last built: 2020-09-01 18:16:49
| Parameter | Value |
|---|---|
| hashtag | #gi2019 |
| start_day | 2019-11-06 |
| end_day | 2019-11-09 |
| timezone | America/New_York |
| theme | theme_light |
| accent | #005190 |
| accent2 | #7FA8C7 |
| kcore | 5 |
| topics_k | 9 |
| bigram_filter | 5 |
| fixed | TRUE |
| seed | 1 |
Introduction
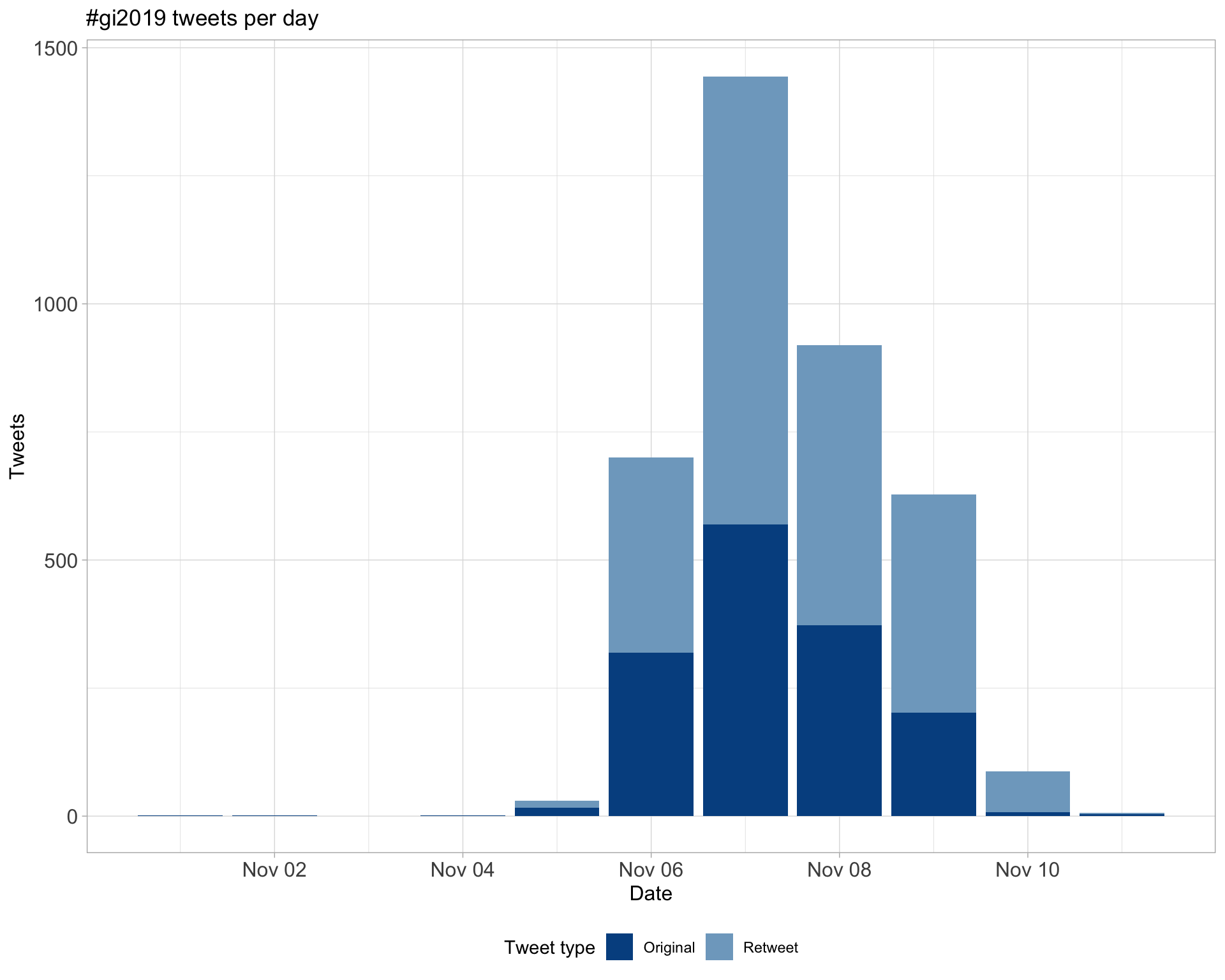
An analysis of tweets from the #gi2019 hashtag for the [Genome Informatics conference][GI2019], 6-9 November 2019, at Cold Spring Harbor Laboratories, New York, USA.
A total of 3820 tweets from 1053 users were collected using the rtweet R package.
1 Timeline
1.1 Tweets by day
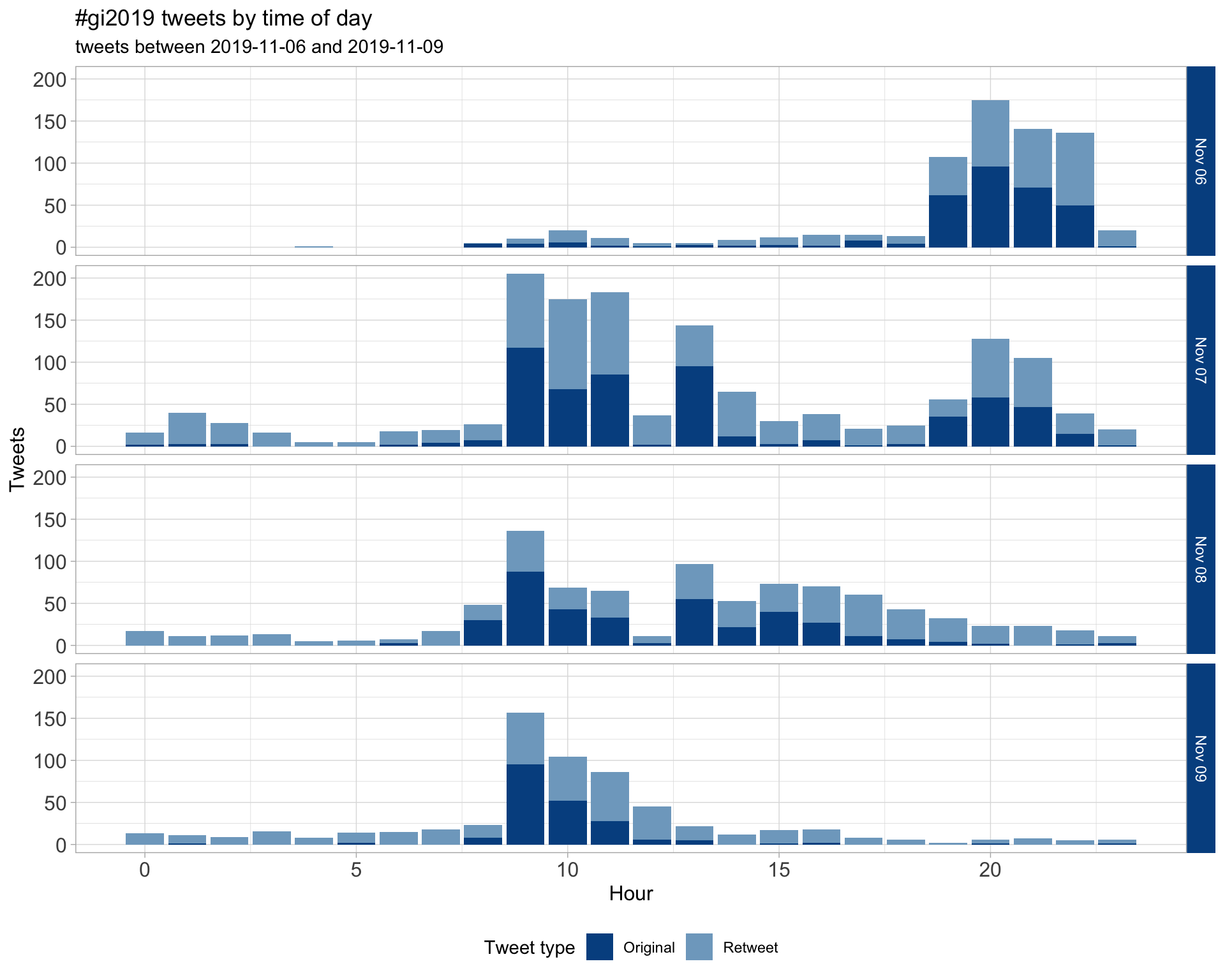
1.2 Tweets by day and time
Filtered for dates 2019-11-06 - 2019-11-09 in the America/New_York timezone.
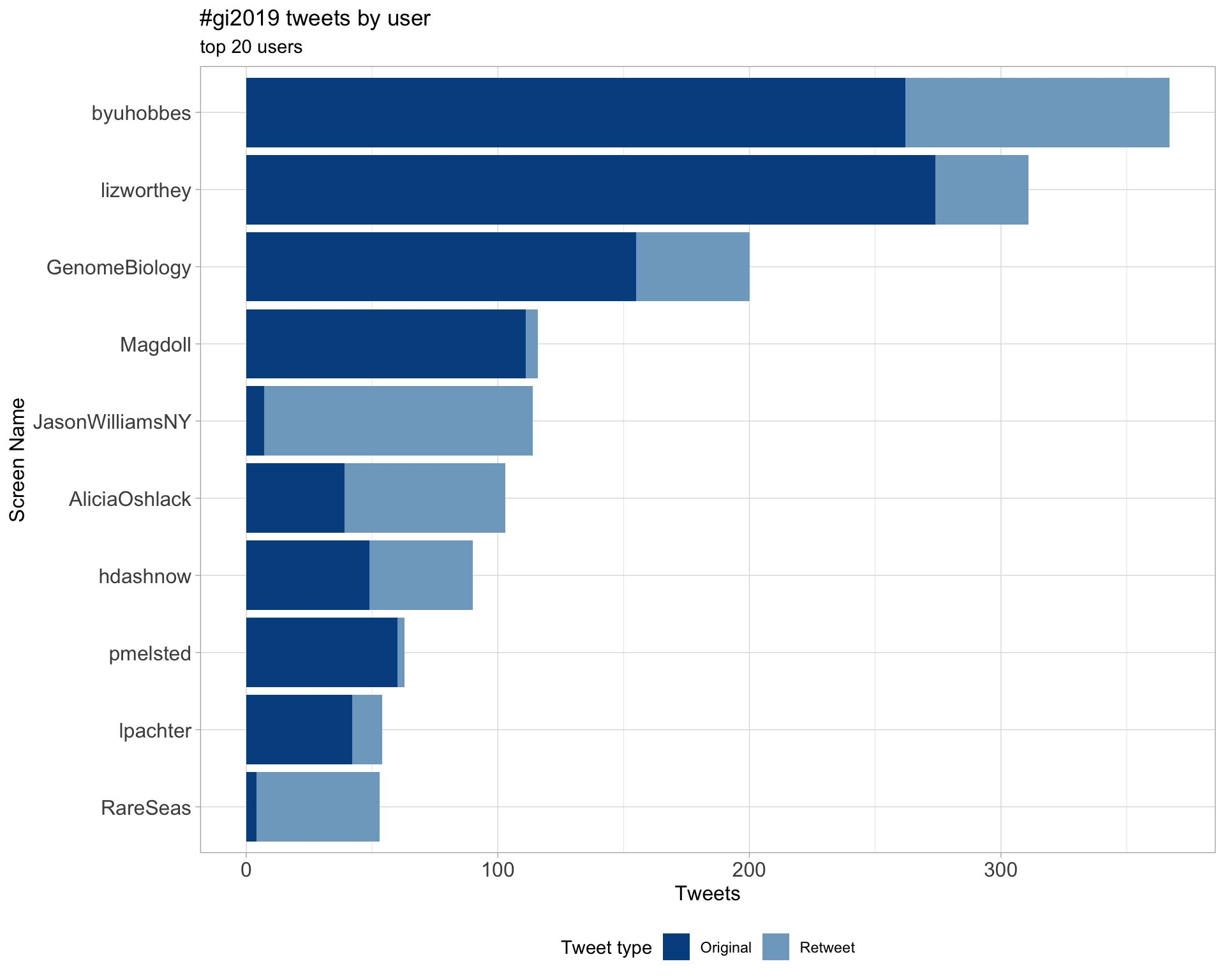
2 Users
2.1 Top tweeters
Overall
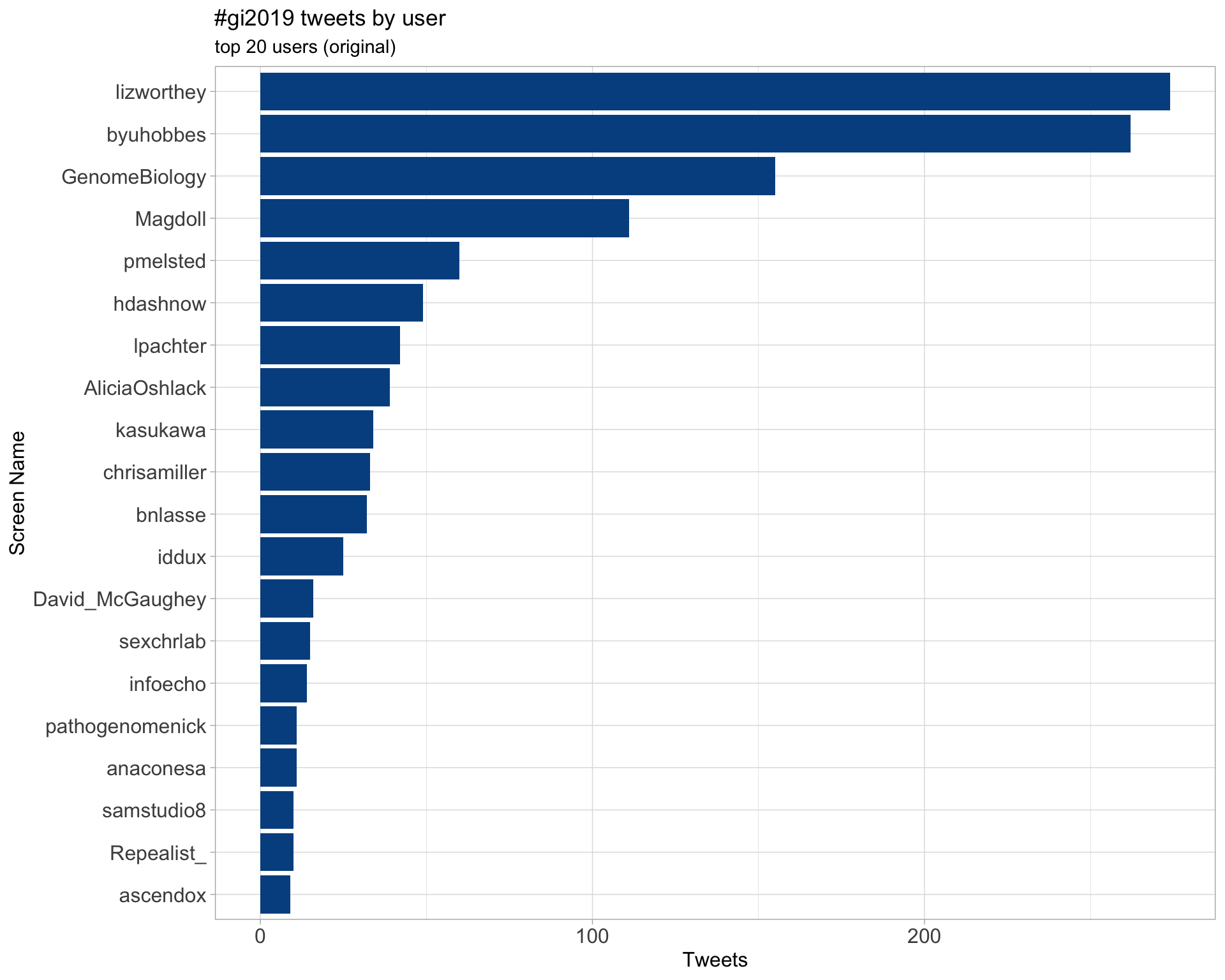
Original
Retweets
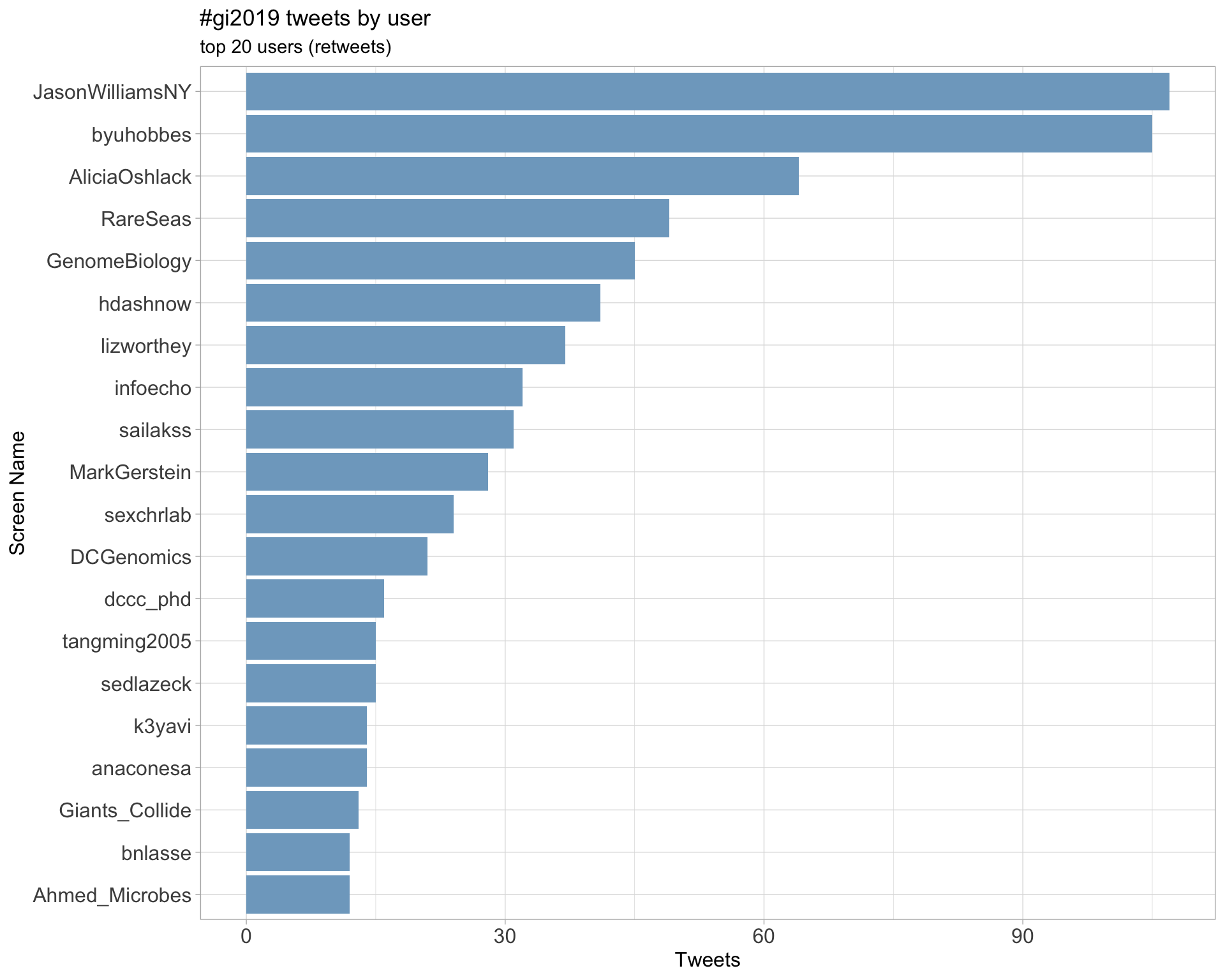
2.2 Retweet proportion
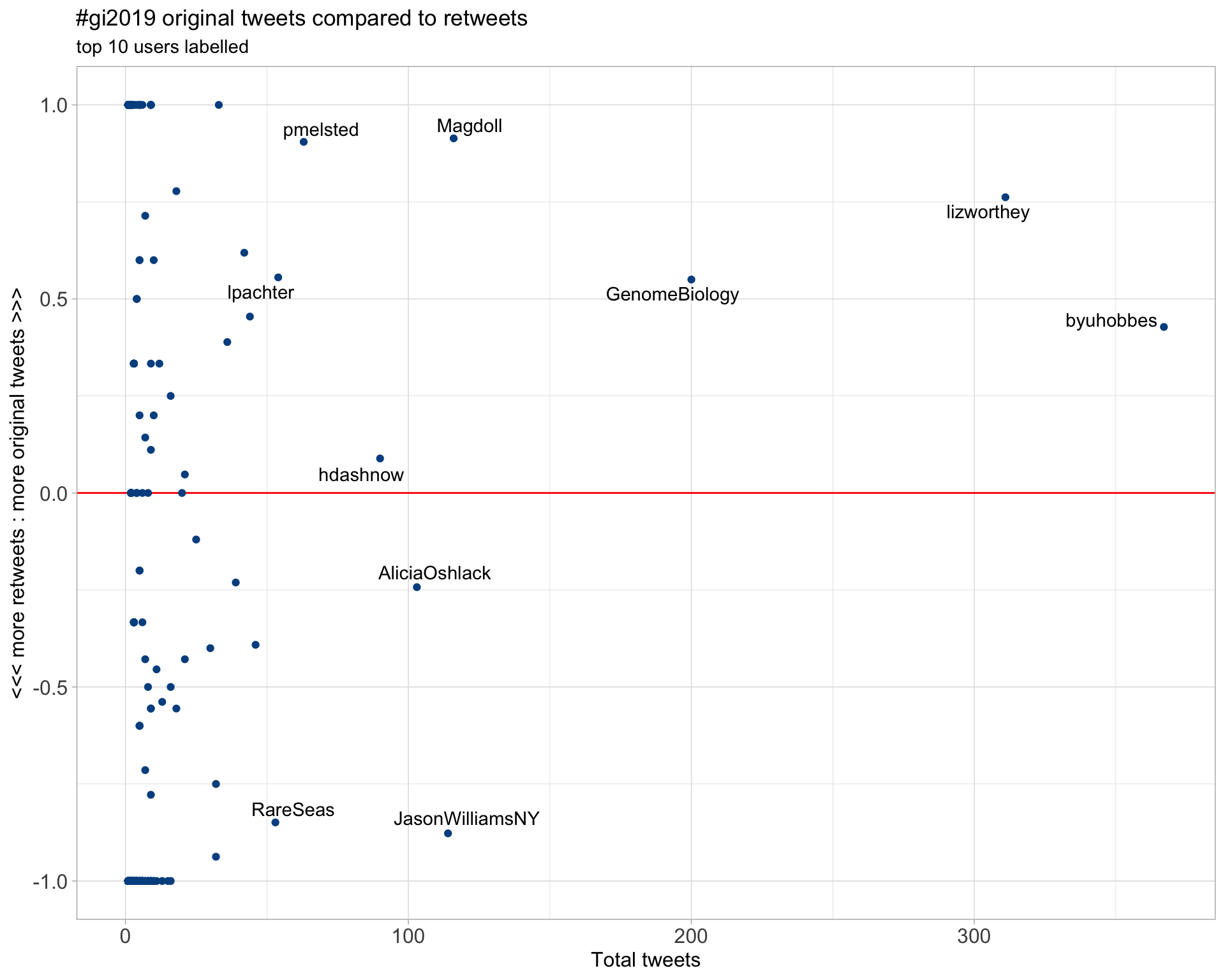
2.3 Top tweeters timeline
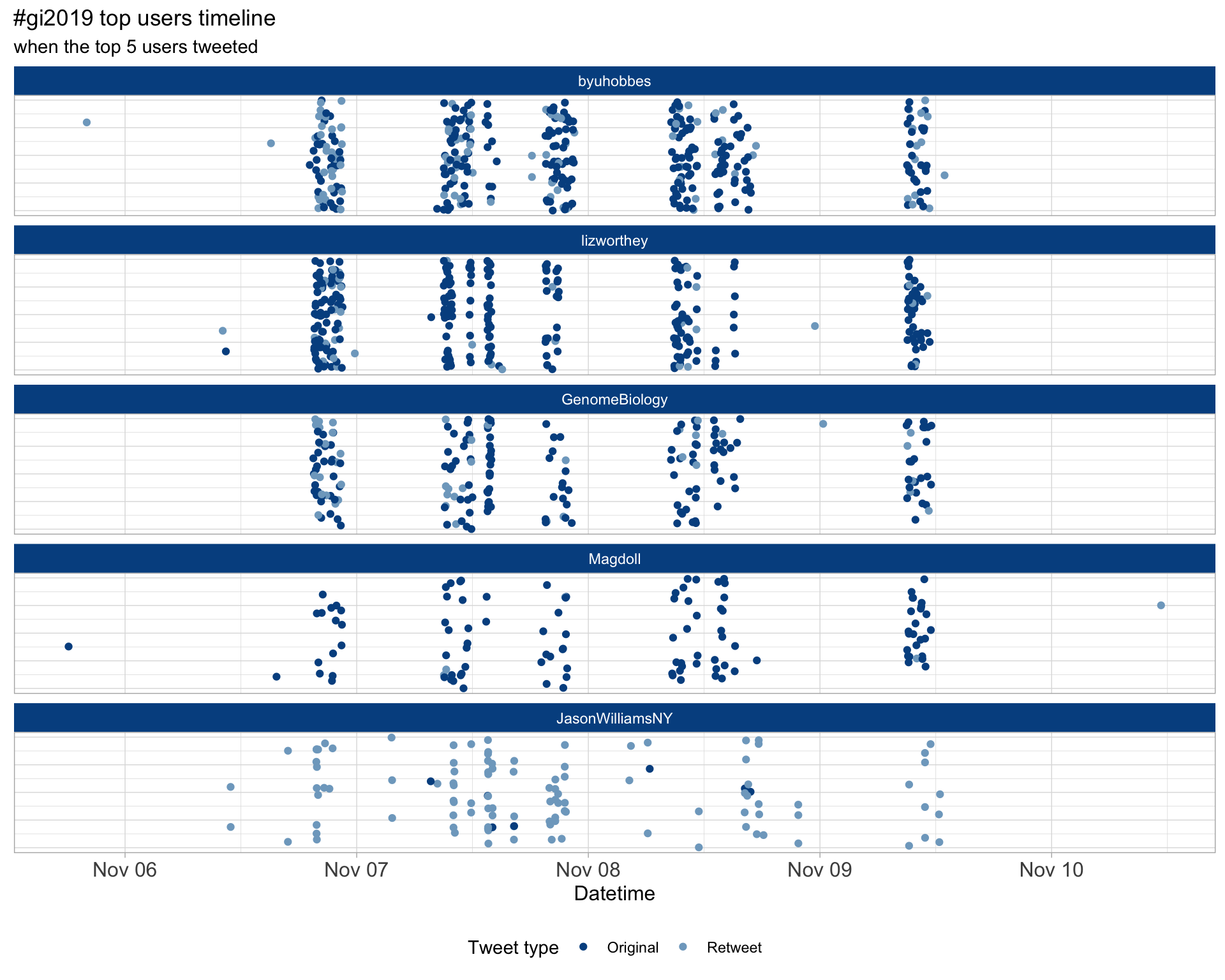
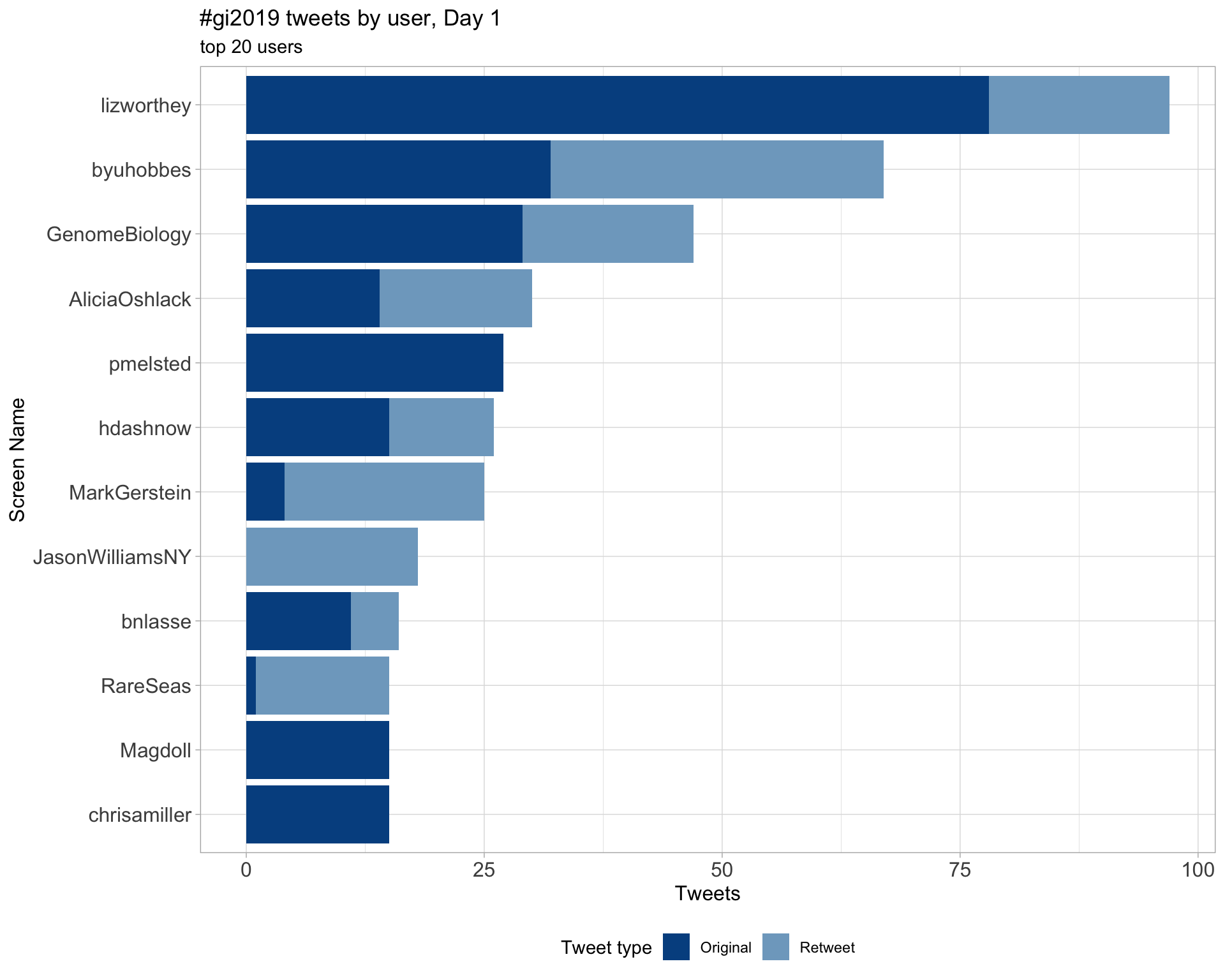
2.4 Top tweeters by day
Overall
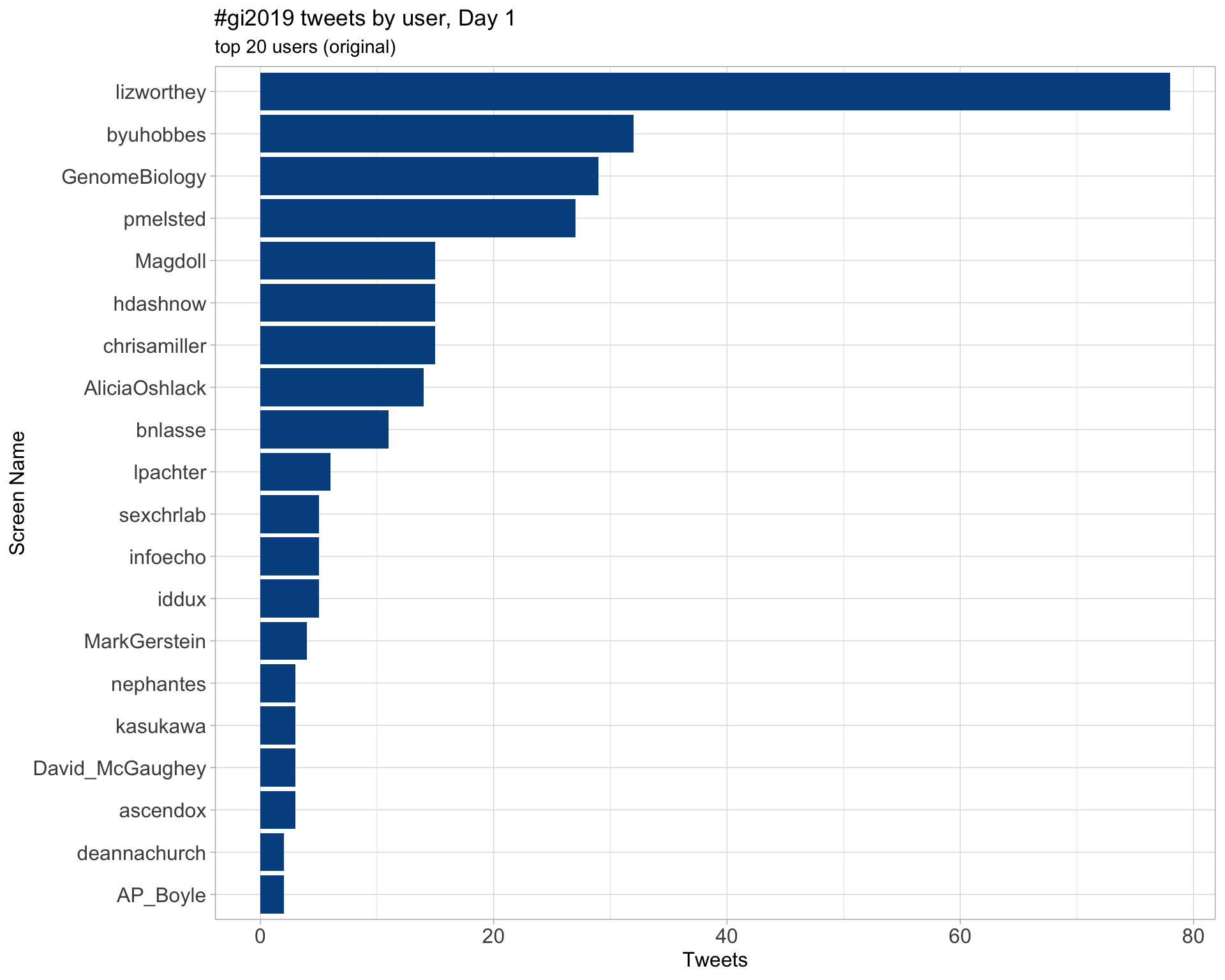
Day 1
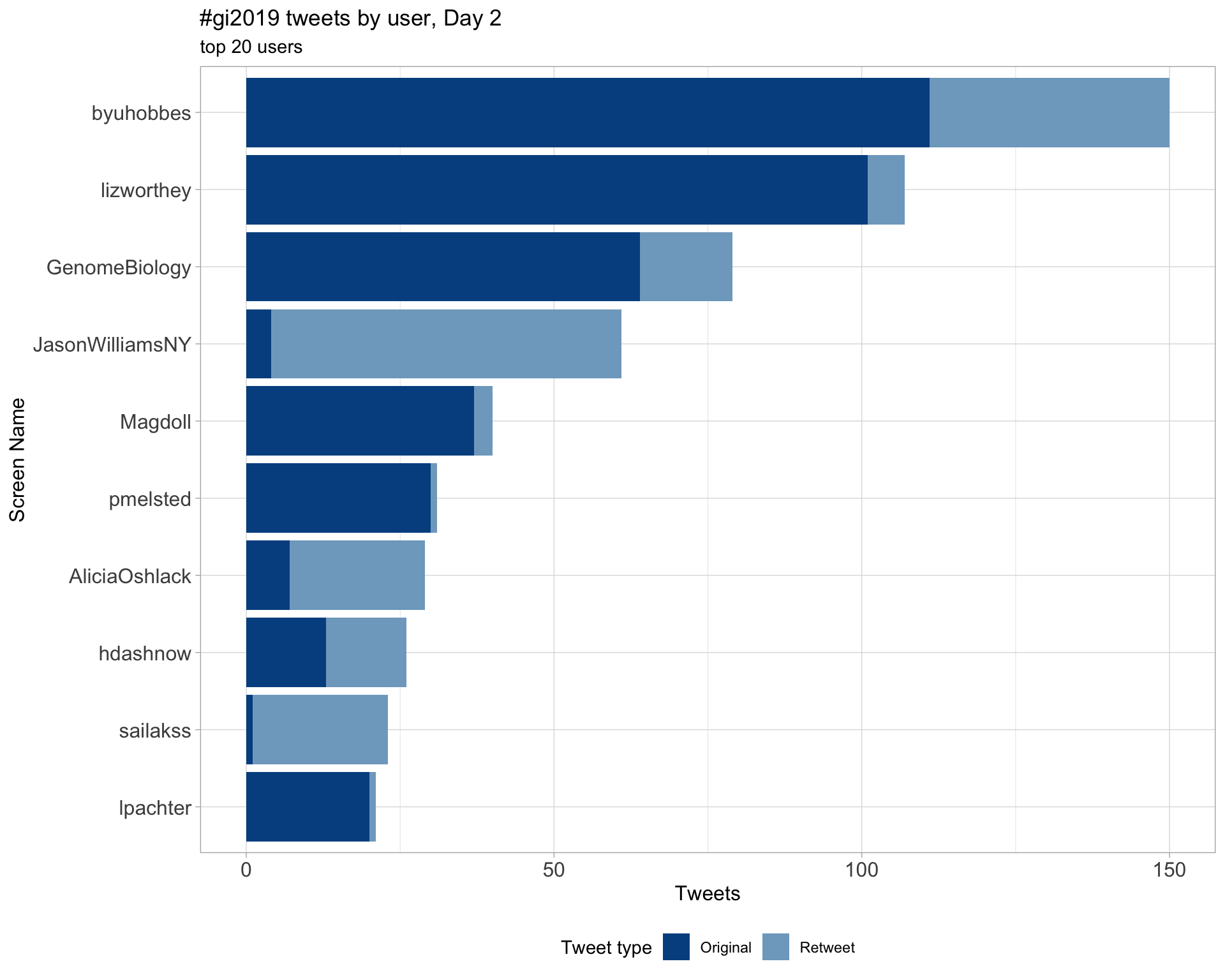
Day 2
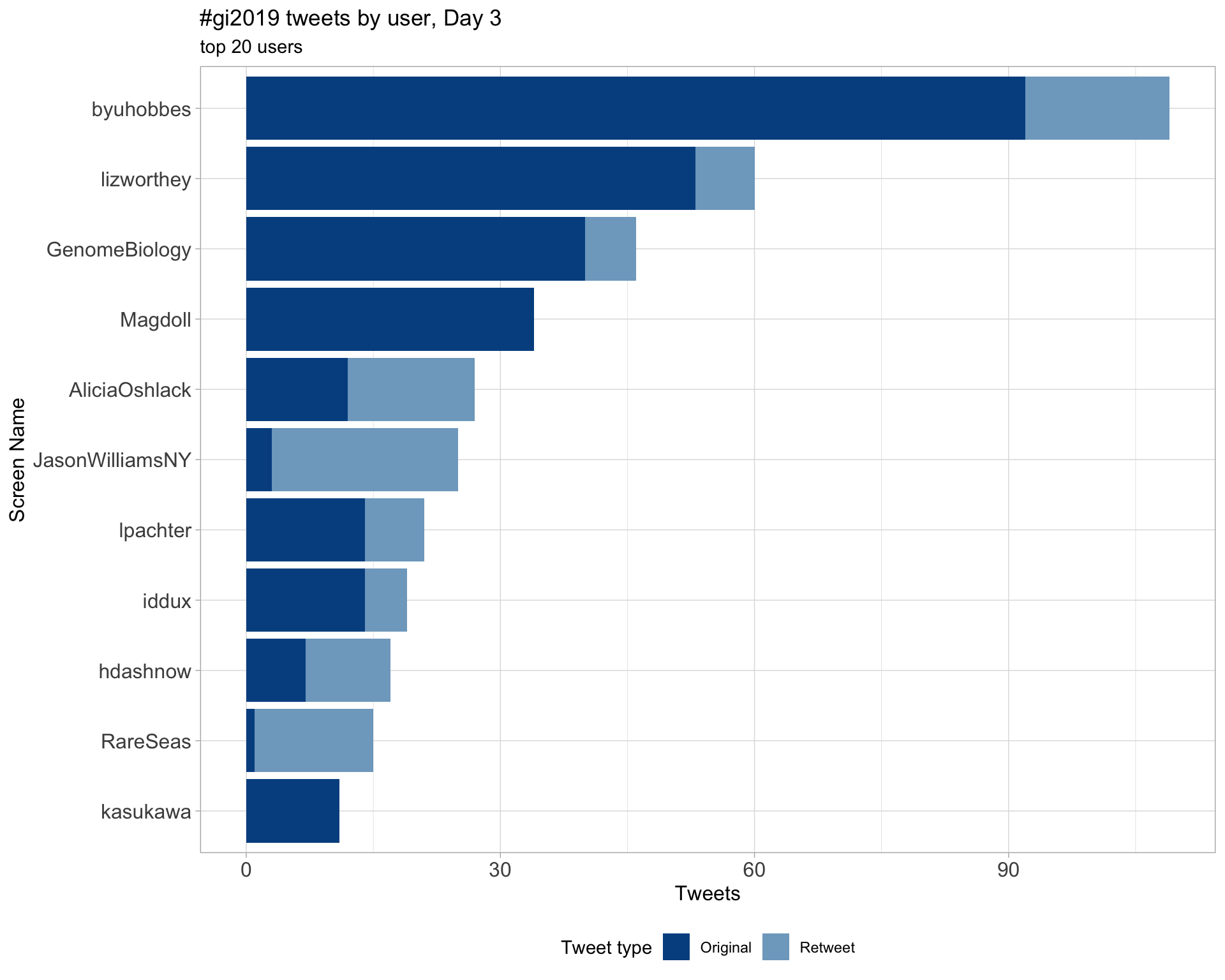
Day 3
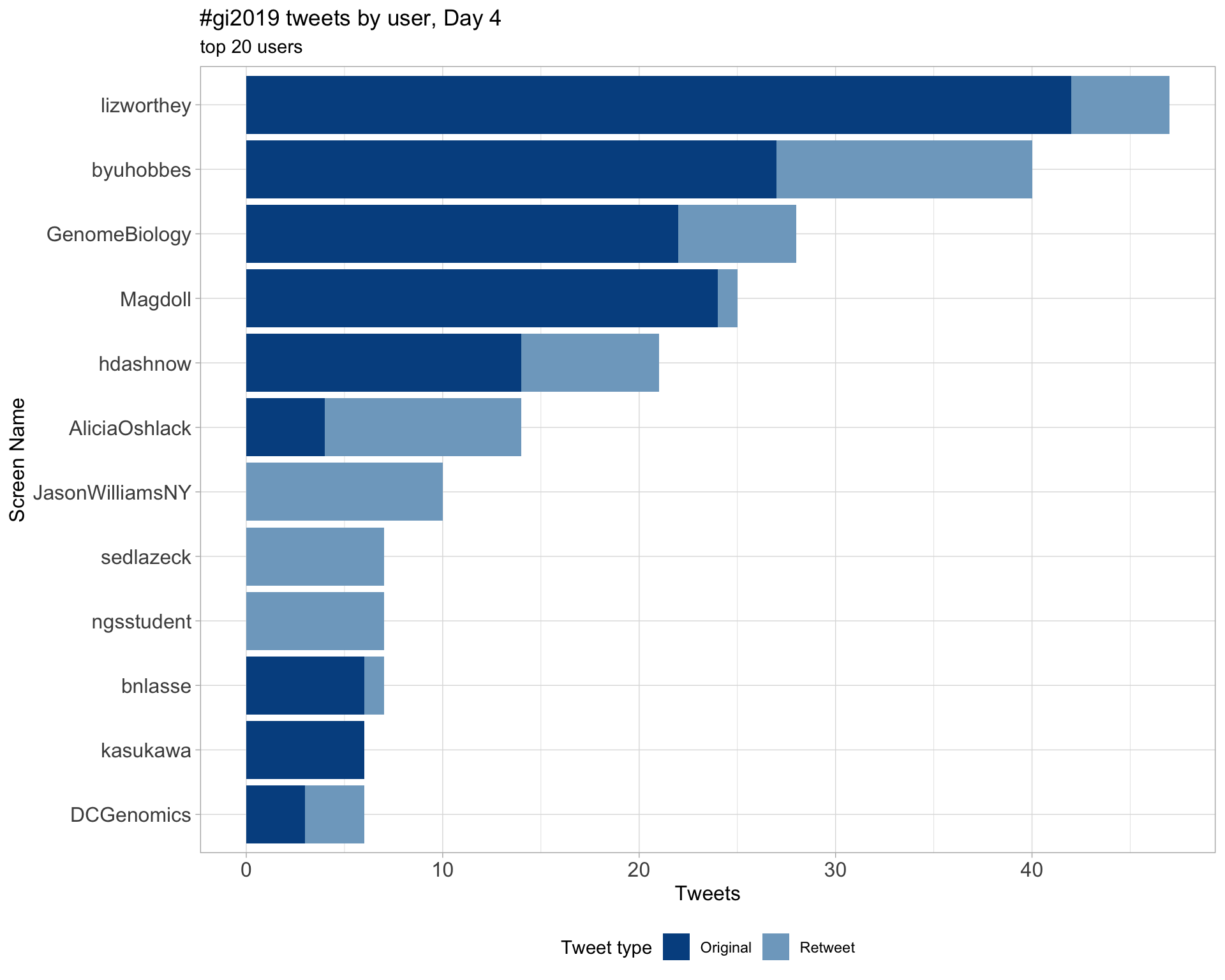
Day 4
Original
Day 1
Day 2
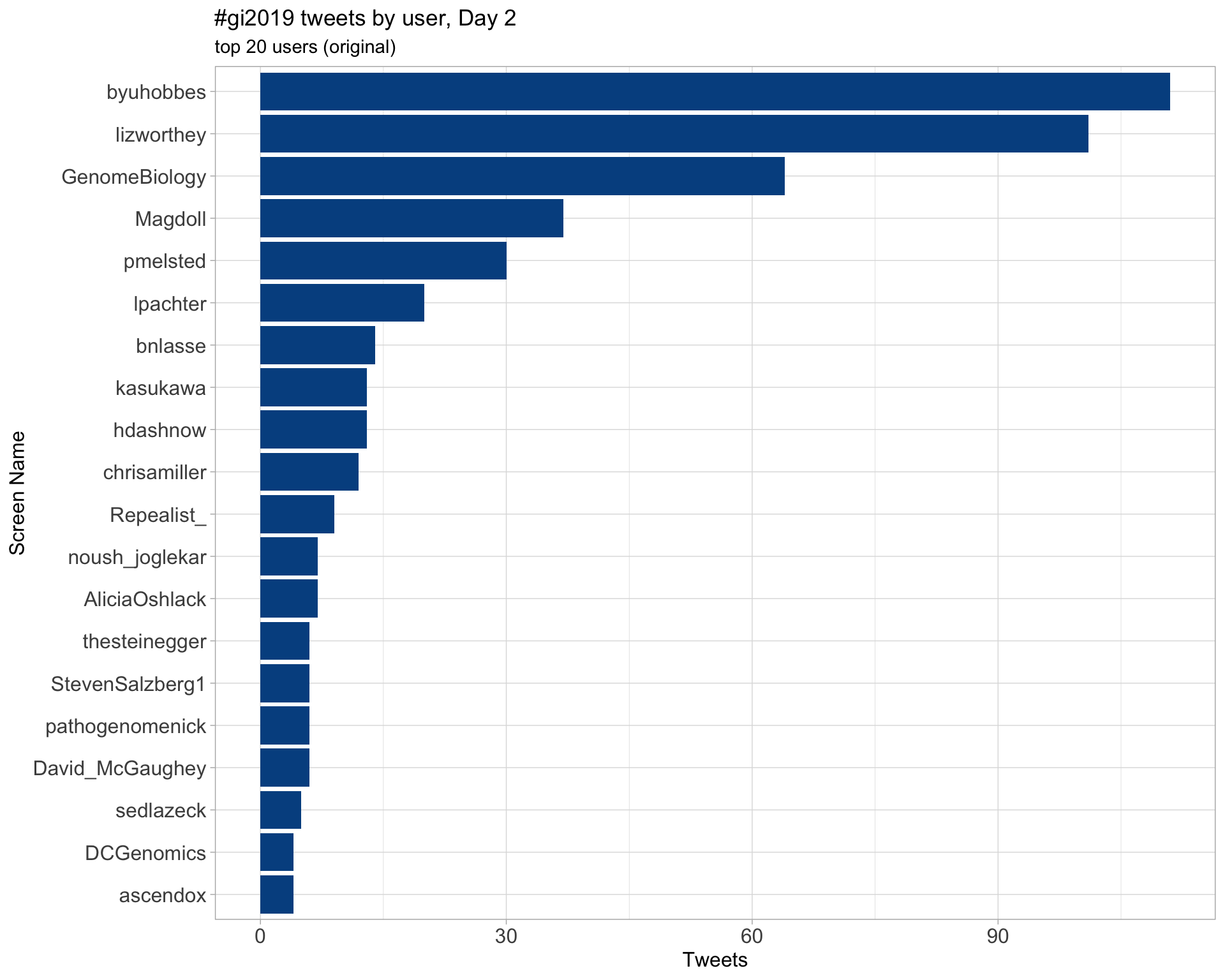
Day 3
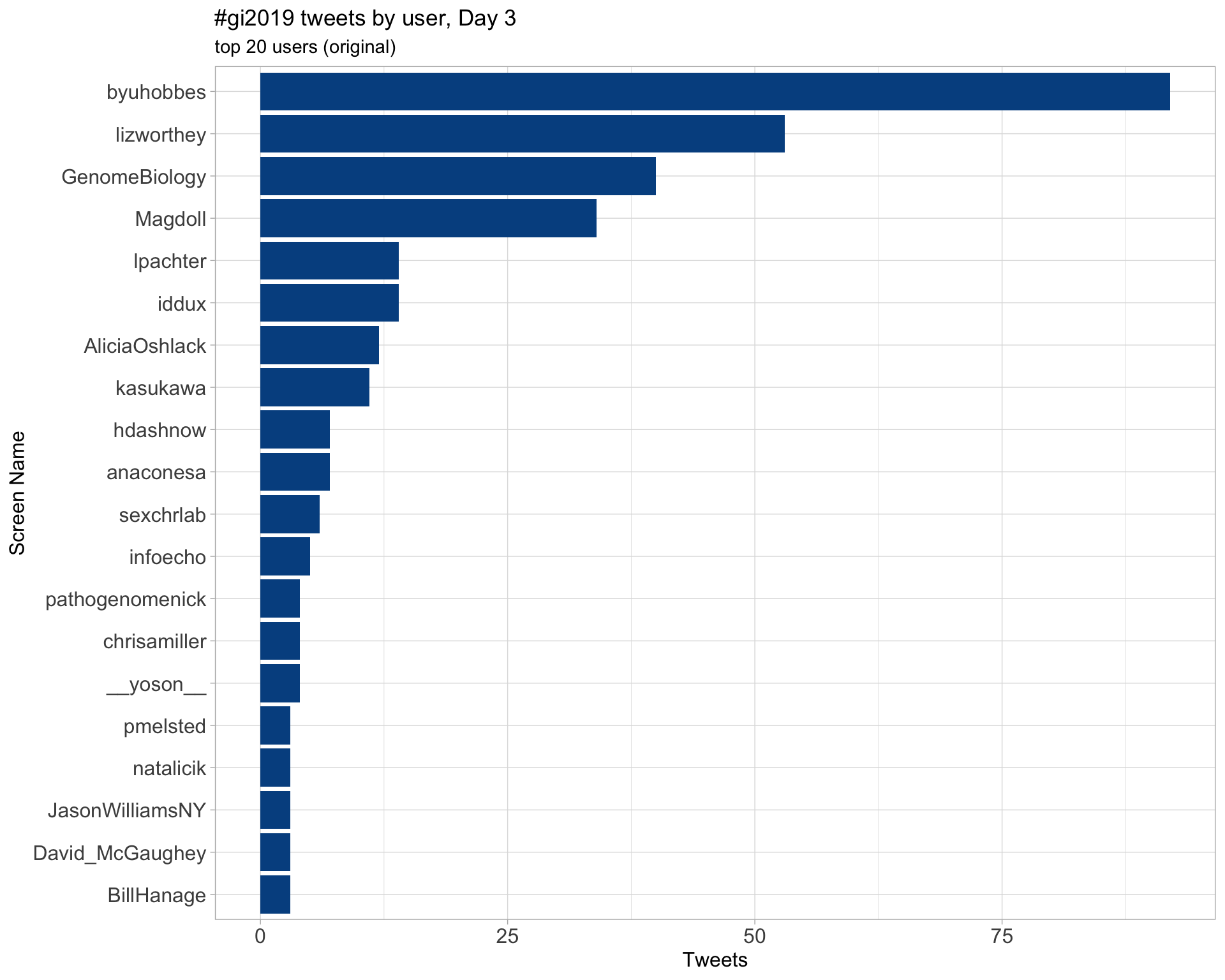
Day 4
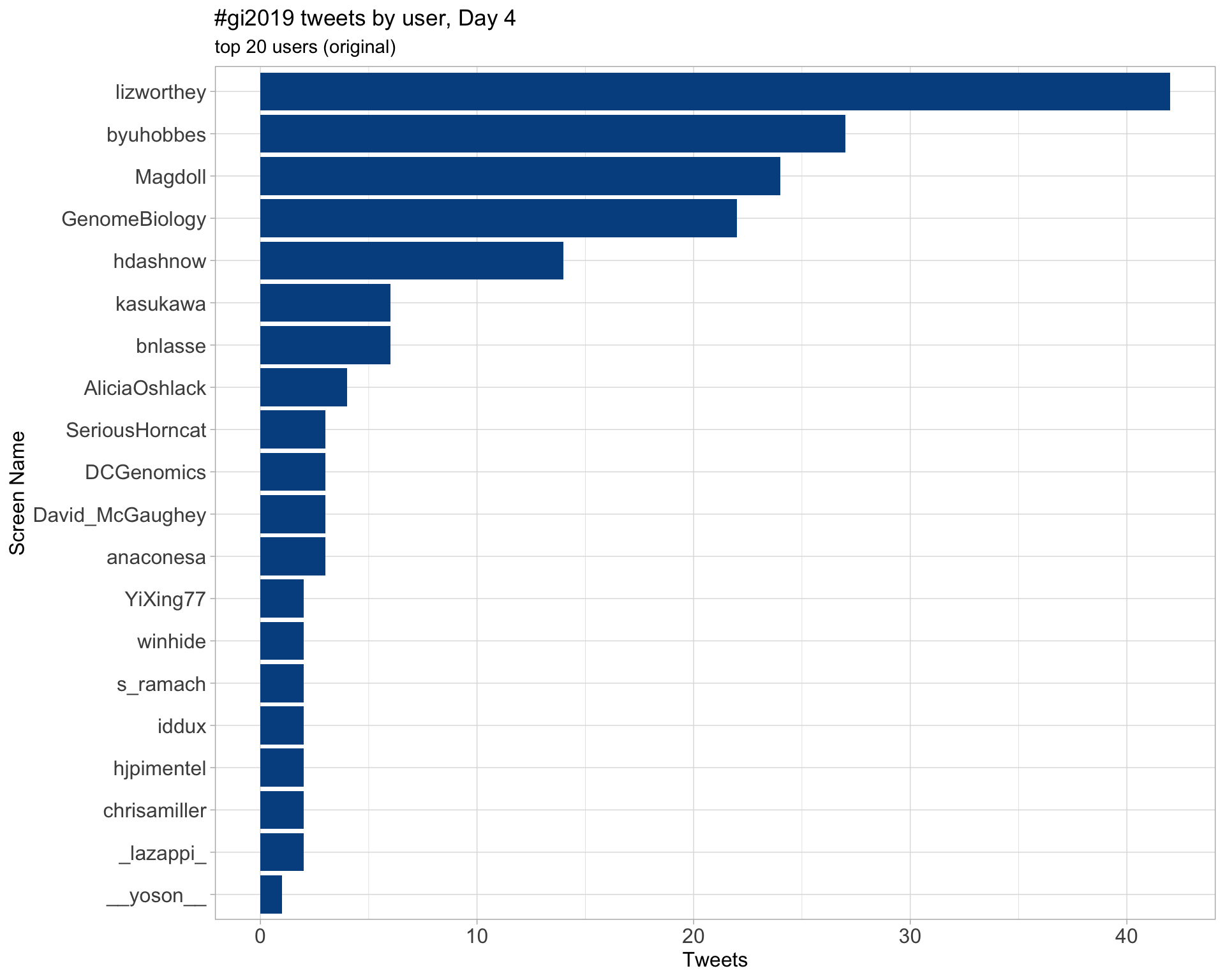
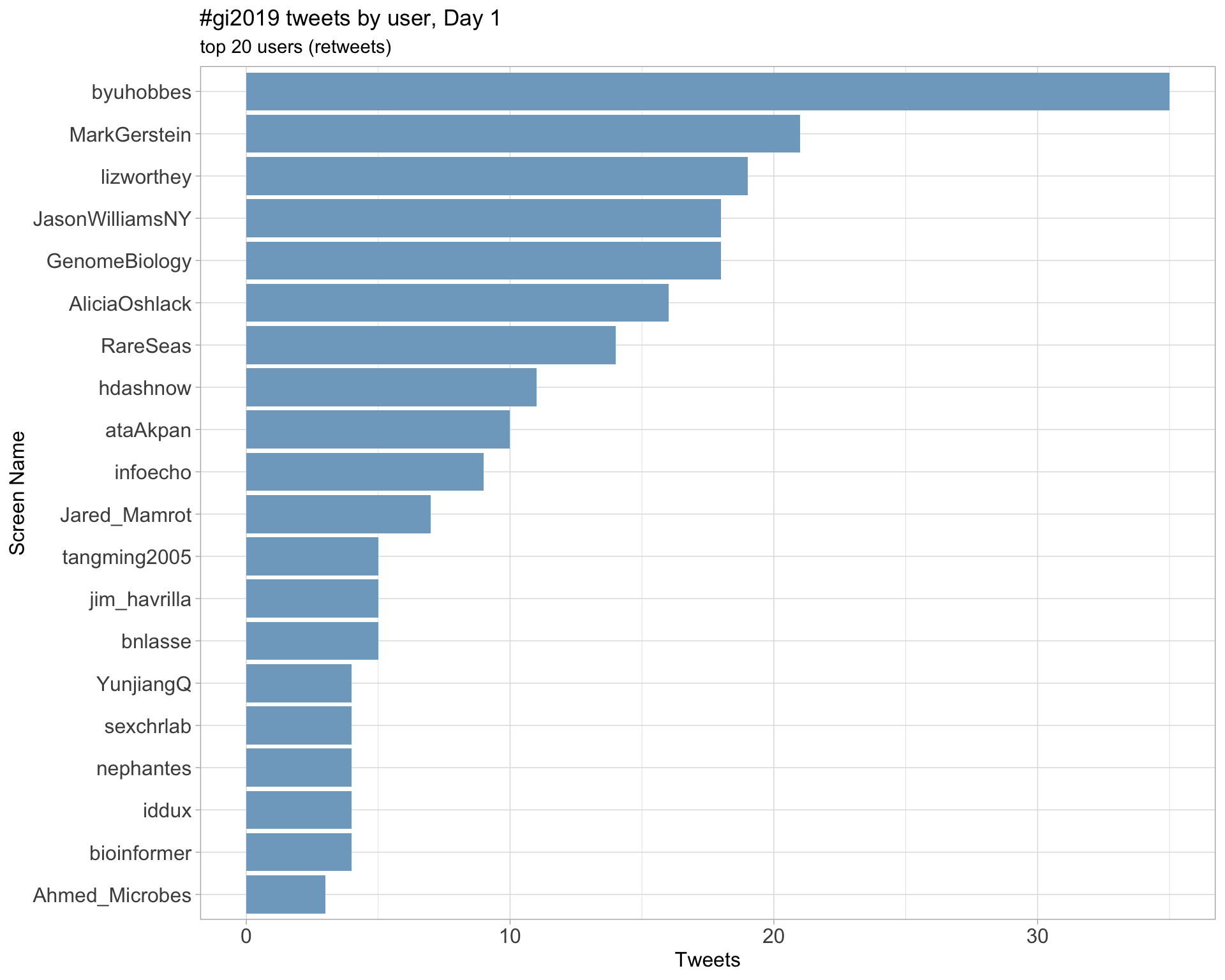
Retweets
Day 1
Day 2
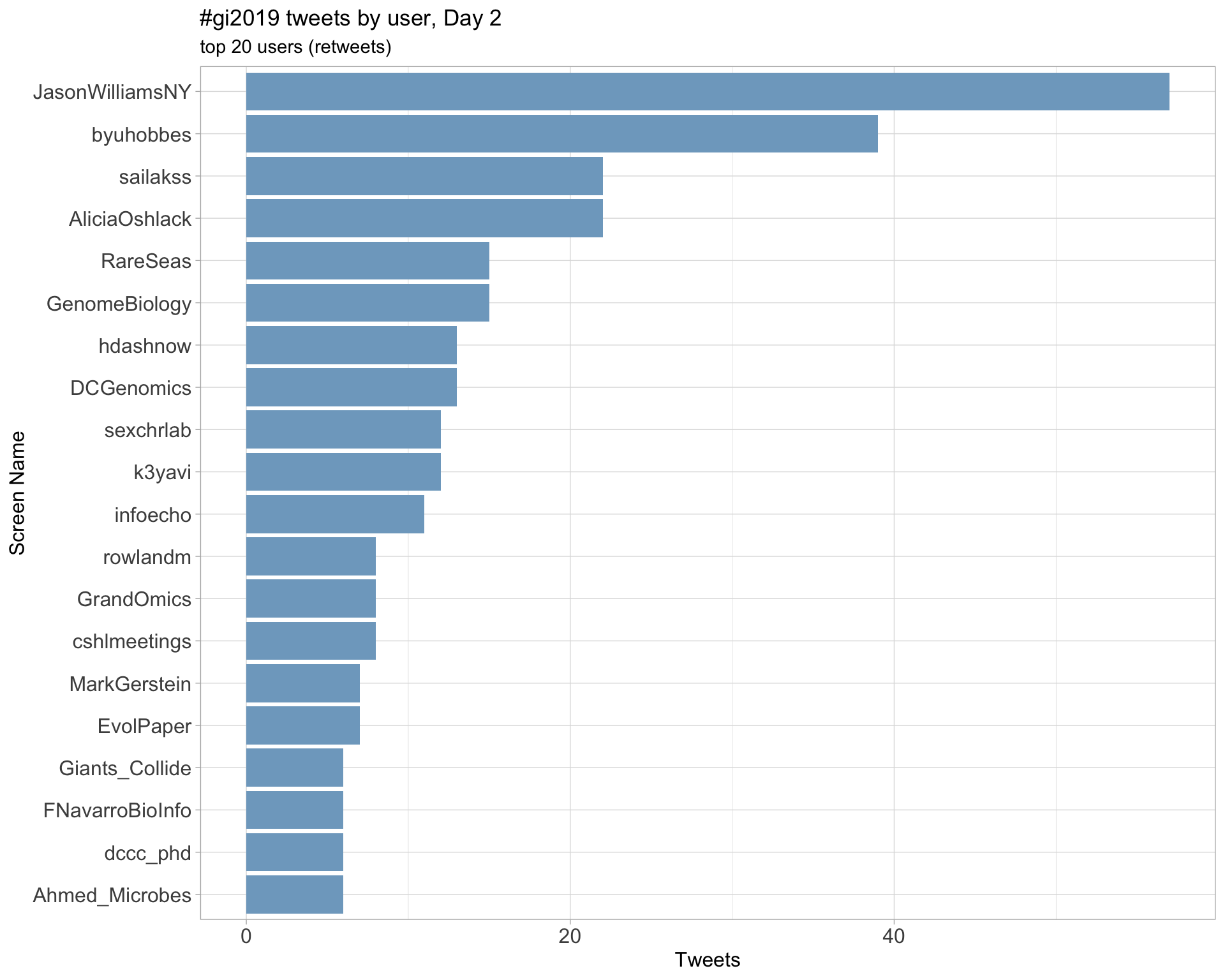
Day 3
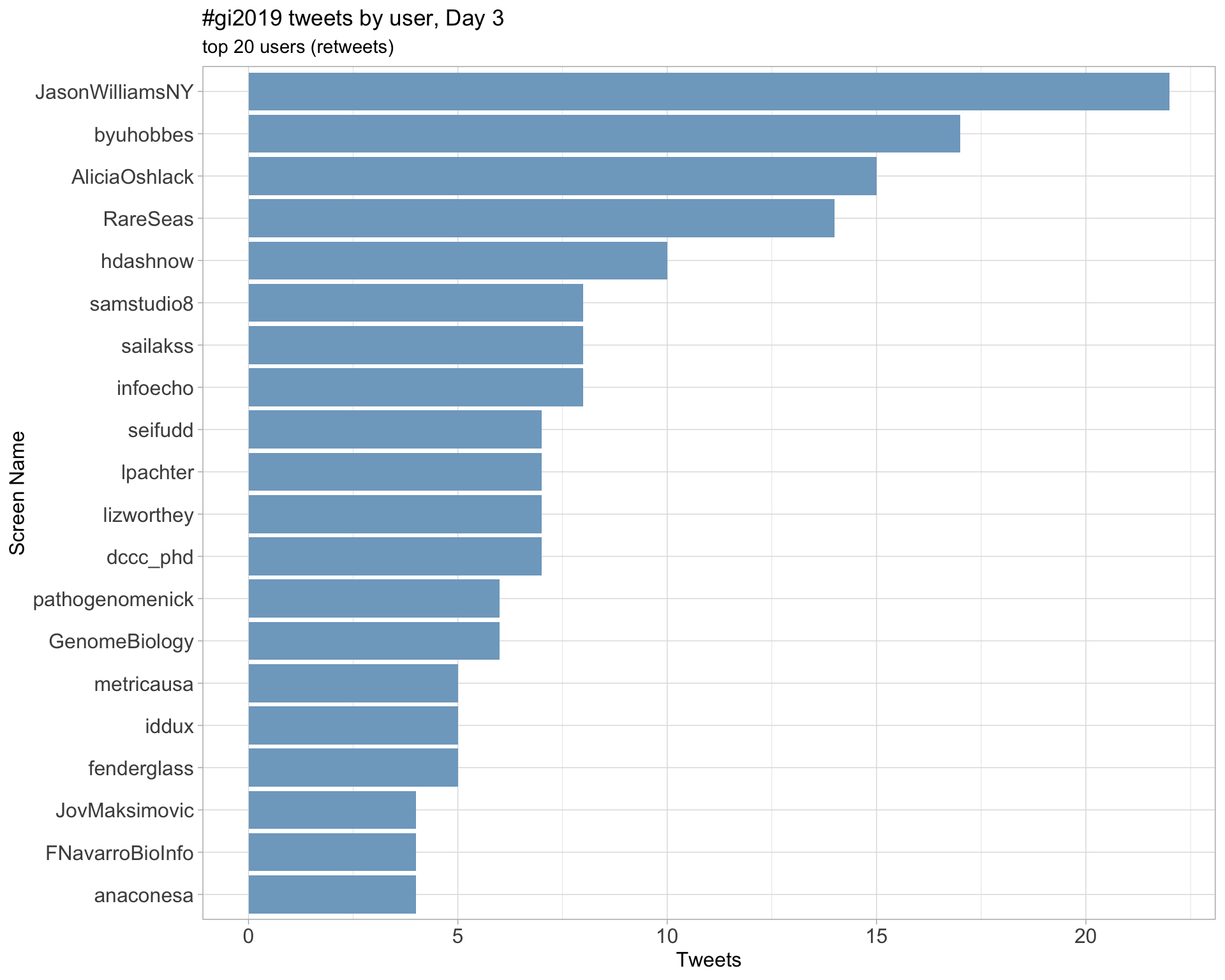
Day 4
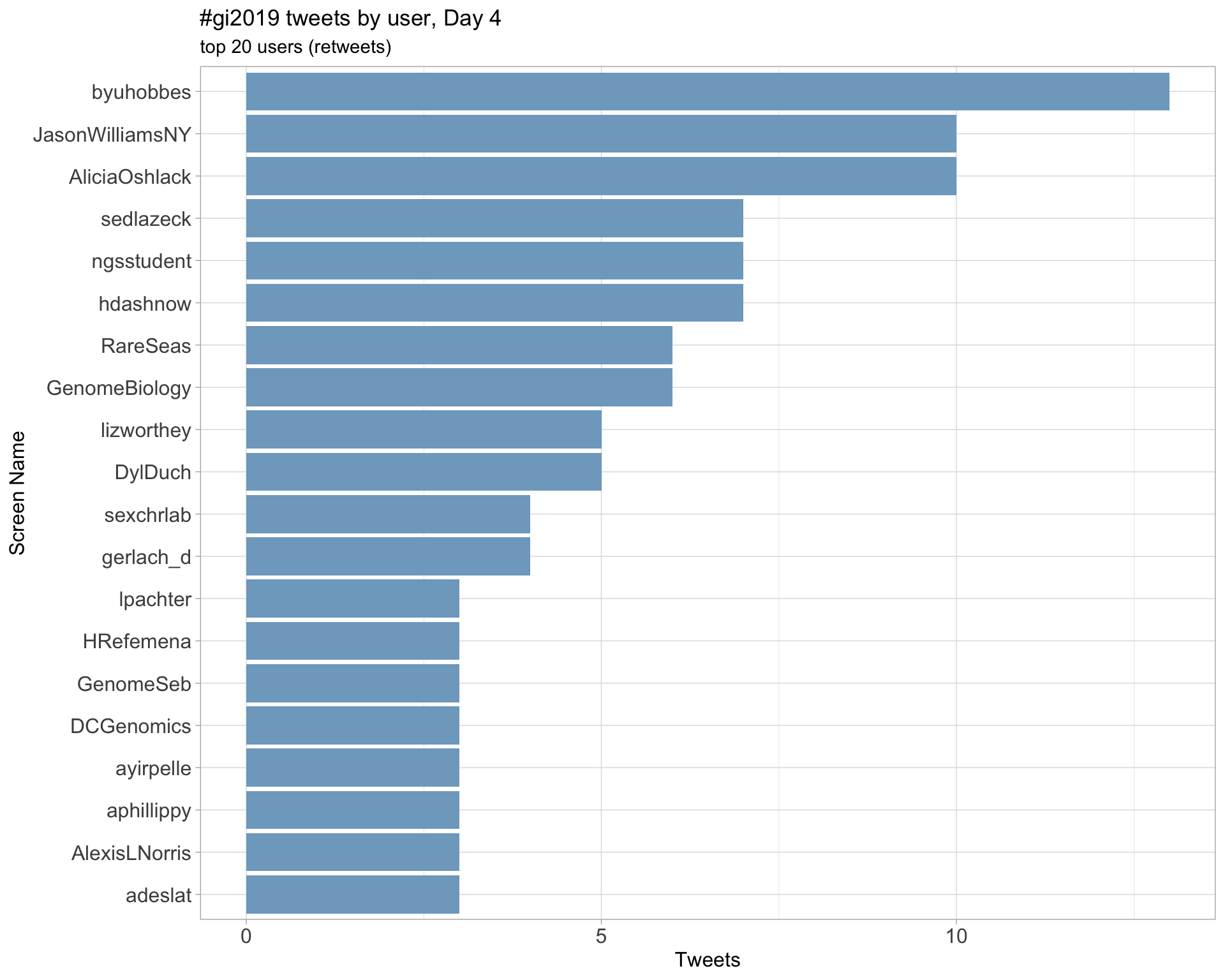
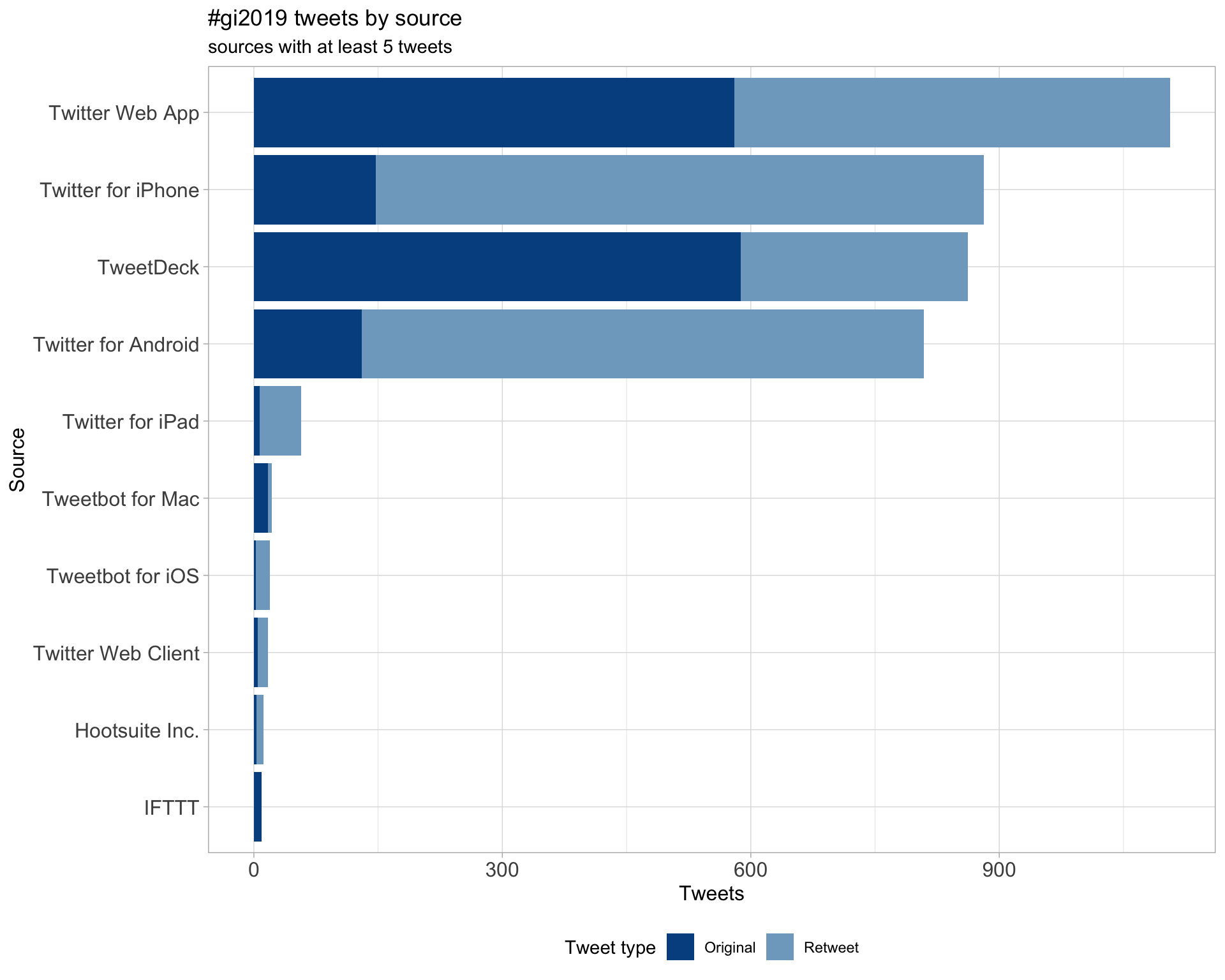
3 Sources
Users
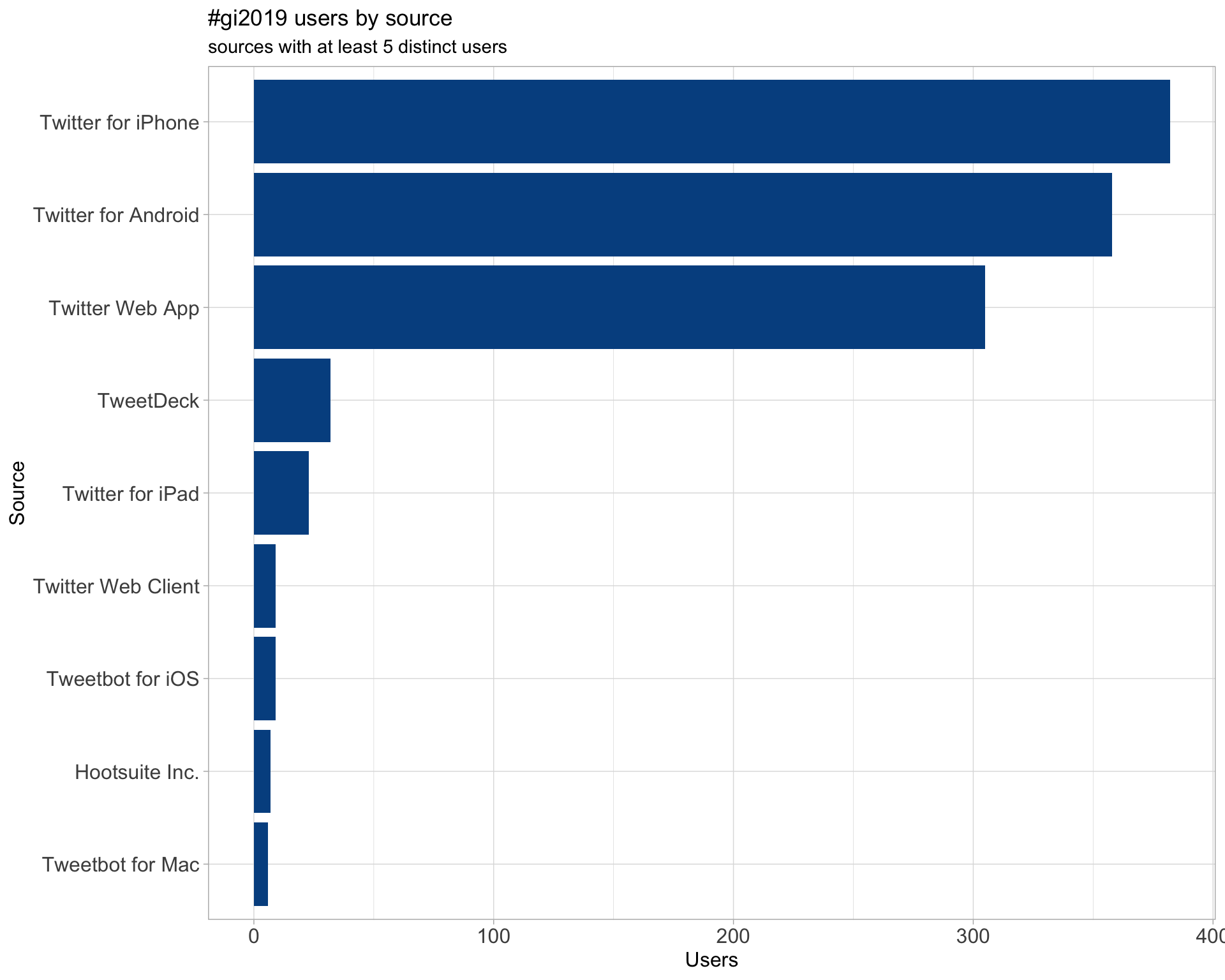
Tweets
4 Networks
4.1 Replies
The “replies network”, composed from users who reply directly to one another, coloured by PageRank.
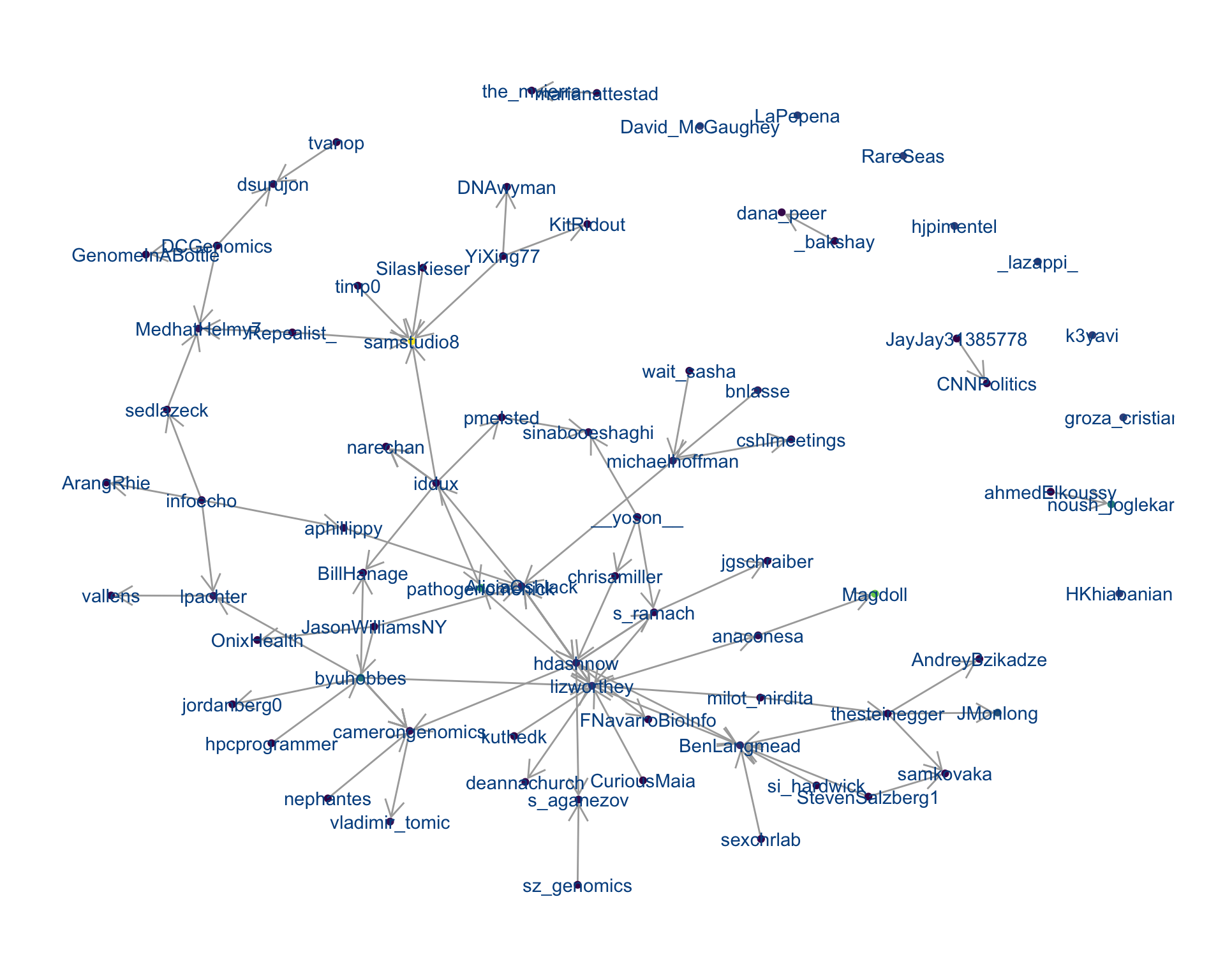
4.2 Mentions
The “mentions network”, where users mention other users in their tweets. Filtered for a k-core of 5. Node colour and size adjusted according to PageRank score.

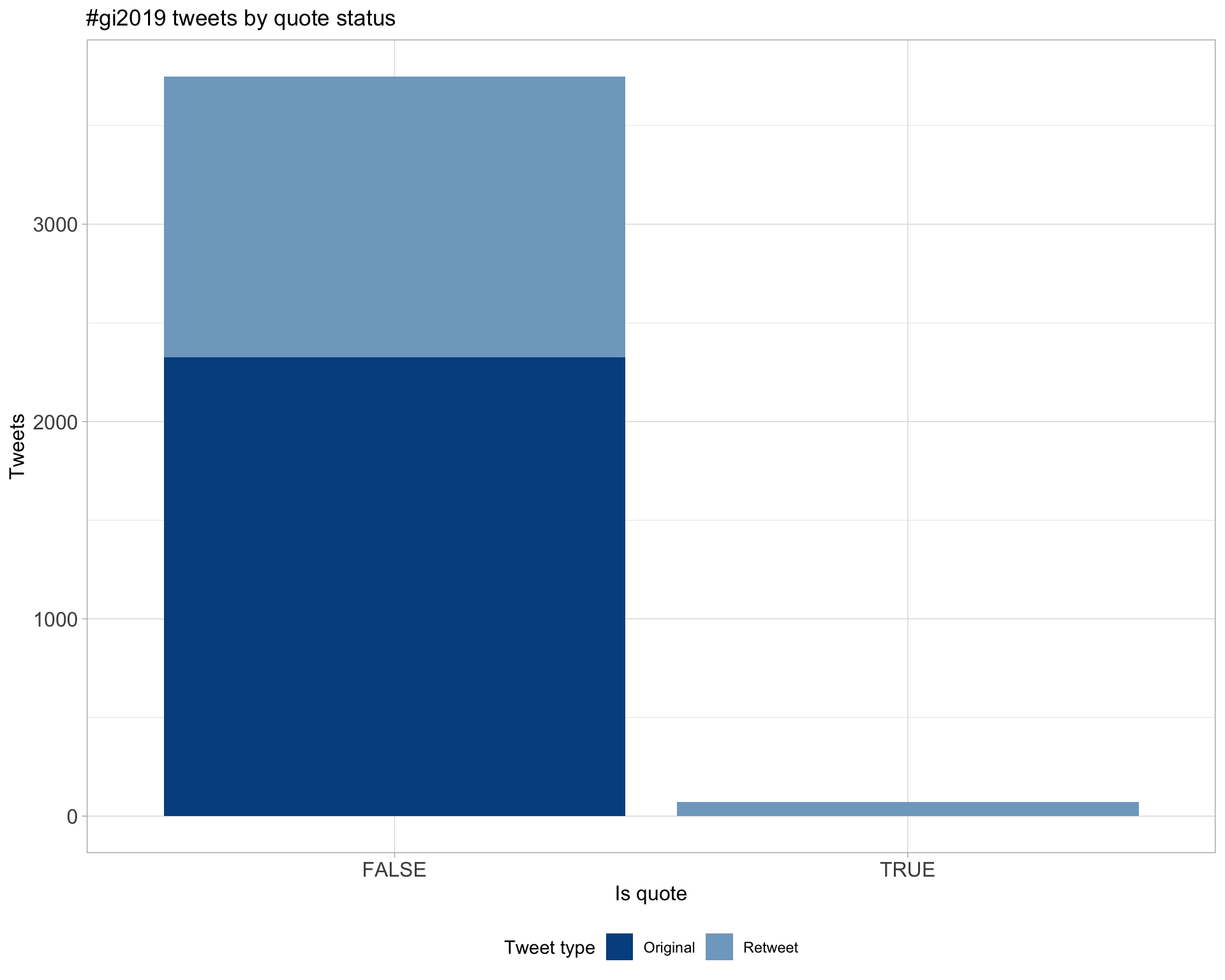
5 Tweet types
5.1 Retweets
Proportion
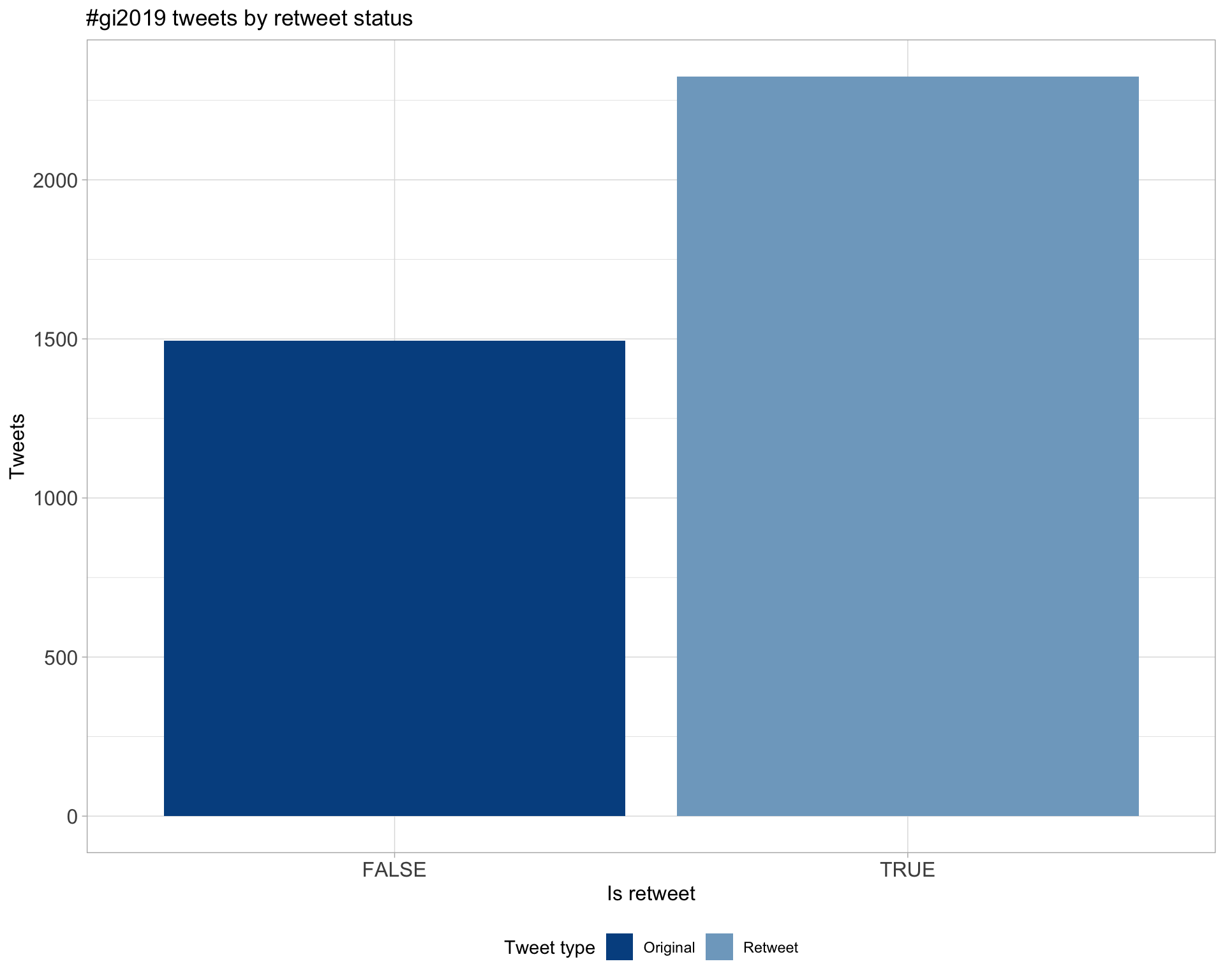
Count
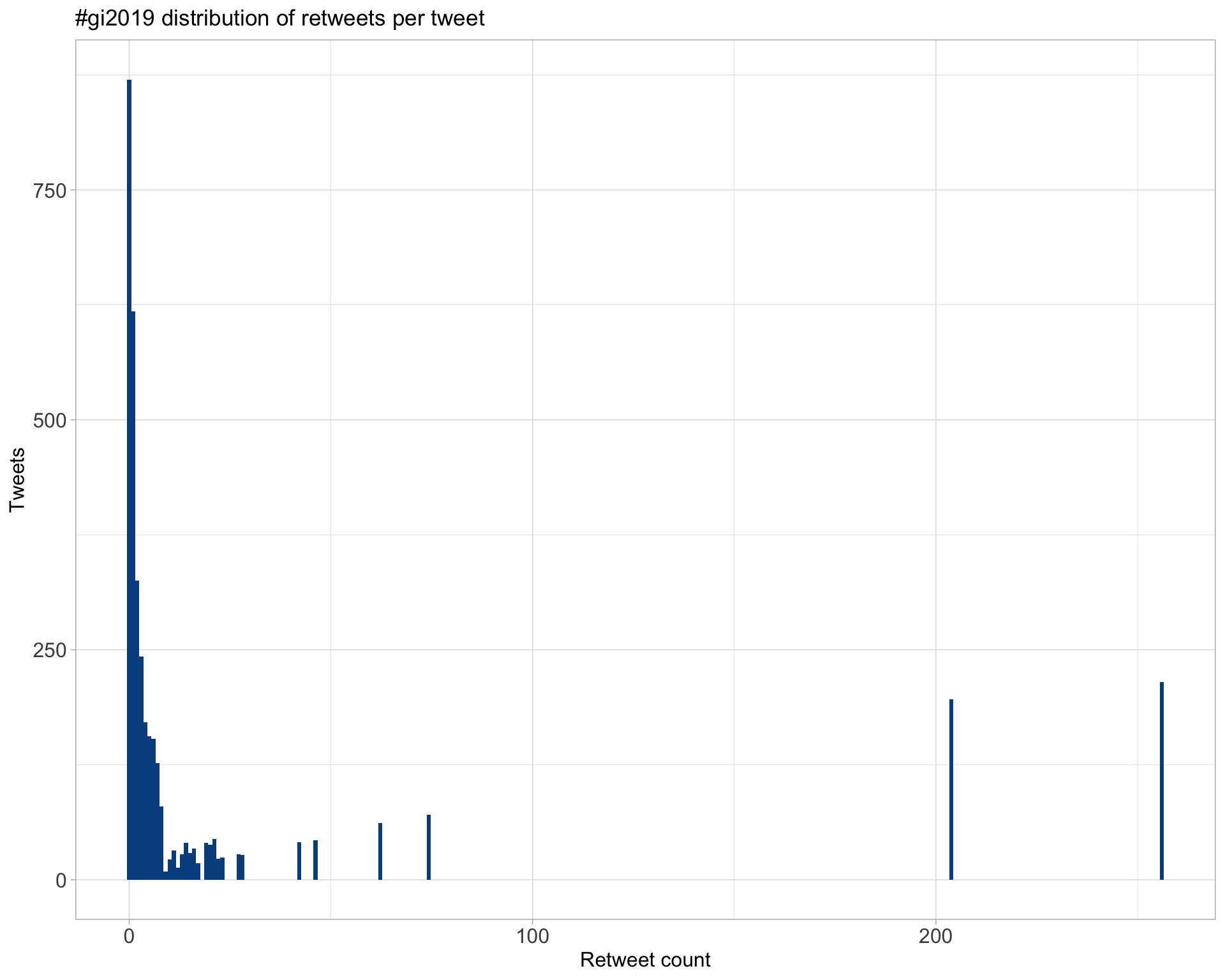
Top 10
| screen_name | text | retweet_count |
|---|---|---|
| lpachter | I highly recommend “How to read (single-cell RNA-seq) PCA plots” (by @vallens): https://t.co/Co3A47SZTR #gi2019 He notes that “A particular danger [in interpreting T-shapes] is that it is tempting to interpret this as a bifurcation in the data.” (it almost never is). #gi2019 | 256 |
| lh3lh3 | I am looking for one or two postdocs working on bioinformatics and computational biology in general. Find me at #gi2019 if you are around. https://t.co/cZpnhzMAtr | 204 |
| aphillippy | Heng is amazing. Go work for him. If he turns you down, come talk to me. I am also looking for students and postdocs at #gi2019 😆 https://t.co/uuuMq6gXLJ | 74 |
| ACSCevents | Save the date - Genome Informatics 2020 - 14-17 Sept at the genome campus #GI2020 #gi2019 https://t.co/EfCc74qrCR | 62 |
| lpachter | A reminder: “cluster sizes in a t-SNE plot mean nothing” https://t.co/GUe2BTFLEO #gi2019 | 46 |
| hdashnow | I’m presenting STRling, our new method to detect novel (and reference) STR expansions from short-read data. https://t.co/hPyL7WBhkI #gi2019 Big thanks to @brent_p who has done a huge amount of work on this algorithm and is an all around fantastic guy to work with. | 42 |
| BenLangmead |
Slides from my #gi2019 talk this morning, about work primarily by @dnb_hopkins: https://t.co/lZYnBK8dhm Software: https://t.co/JALMMmfSca Library: https://t.co/8Yifw0NUsz |
28 |
| deannachurch | Also- I’m looking for someone to lead my Informatics/analysis group- and single cell experience is a must! Help build the next generation of genomics tools using genome engineering! https://t.co/v8vzvssQUM #gi2019 | 27 |
| infoecho | Real bioinformatics “file parsing”!!#gi2019 https://t.co/4vll3qI61u | 23 |
| lpachter | We’ve posted the kallisto | bustools poster presented by @sinabooeshaghi at #gi2019 on @F1000Research https://t.co/EVxAXZvGAC https://t.co/inzS4JAeUZ | 22 |
Most retweeted
5.2 Likes
Proportion
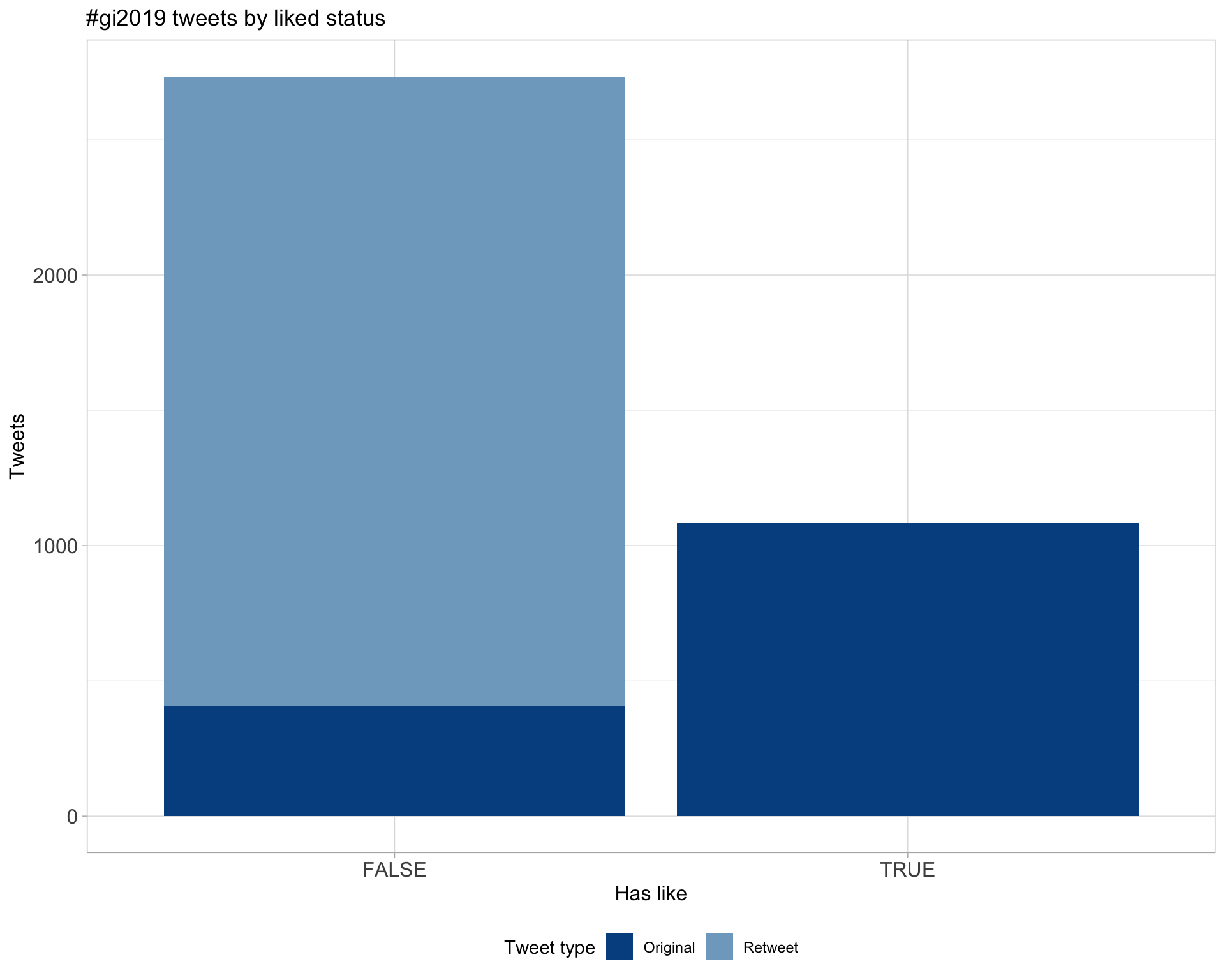
Count
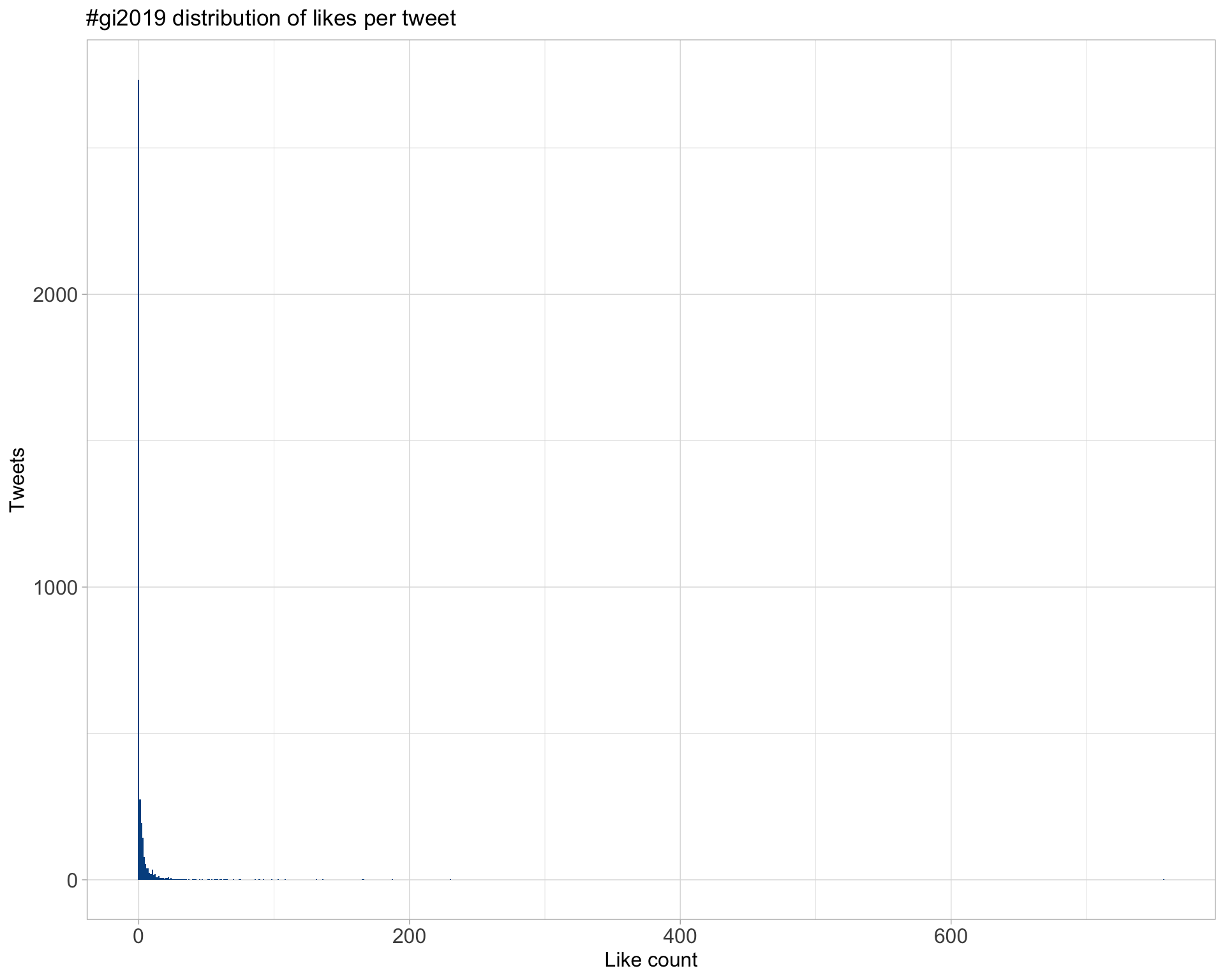
Top 10
| screen_name | text | favorite_count |
|---|---|---|
| lpachter | I highly recommend “How to read (single-cell RNA-seq) PCA plots” (by @vallens): https://t.co/Co3A47SZTR #gi2019 He notes that “A particular danger [in interpreting T-shapes] is that it is tempting to interpret this as a bifurcation in the data.” (it almost never is). #gi2019 | 757 |
| lh3lh3 | I am looking for one or two postdocs working on bioinformatics and computational biology in general. Find me at #gi2019 if you are around. https://t.co/cZpnhzMAtr | 230 |
| samstudio8 | Made it to #gi2019 at @CSHL. My metagenomic assembly graph artwork is the book cover! https://t.co/T2X6EPqweC | 187 |
| lpachter | A reminder: “cluster sizes in a t-SNE plot mean nothing” https://t.co/GUe2BTFLEO #gi2019 | 166 |
| aphillippy | Heng is amazing. Go work for him. If he turns you down, come talk to me. I am also looking for students and postdocs at #gi2019 😆 https://t.co/uuuMq6gXLJ | 165 |
| ACSCevents | Save the date - Genome Informatics 2020 - 14-17 Sept at the genome campus #GI2020 #gi2019 https://t.co/EfCc74qrCR | 136 |
| sexchrlab |
I can’t begin to tell you how much I love the openness of Genome Informatics. I know science has a long way to go, but the culture of sharing data and code, and emphasizing validation; it warms my heart. #GI2019 |
131 |
| infoecho | A Manhattan plot for #GI2019 https://t.co/Jpamfgex8e | 108 |
| hdashnow | I’m presenting STRling, our new method to detect novel (and reference) STR expansions from short-read data. https://t.co/hPyL7WBhkI #gi2019 Big thanks to @brent_p who has done a huge amount of work on this algorithm and is an all around fantastic guy to work with. | 103 |
| chrisamiller |
SN: Not every bioinformatician gets to go to the algorithm ball. Some of them have to stay home and write pipelines Preach, brother 😆 #gi2019 |
98 |
Most likes
5.3 Quotes
Proportion
Count
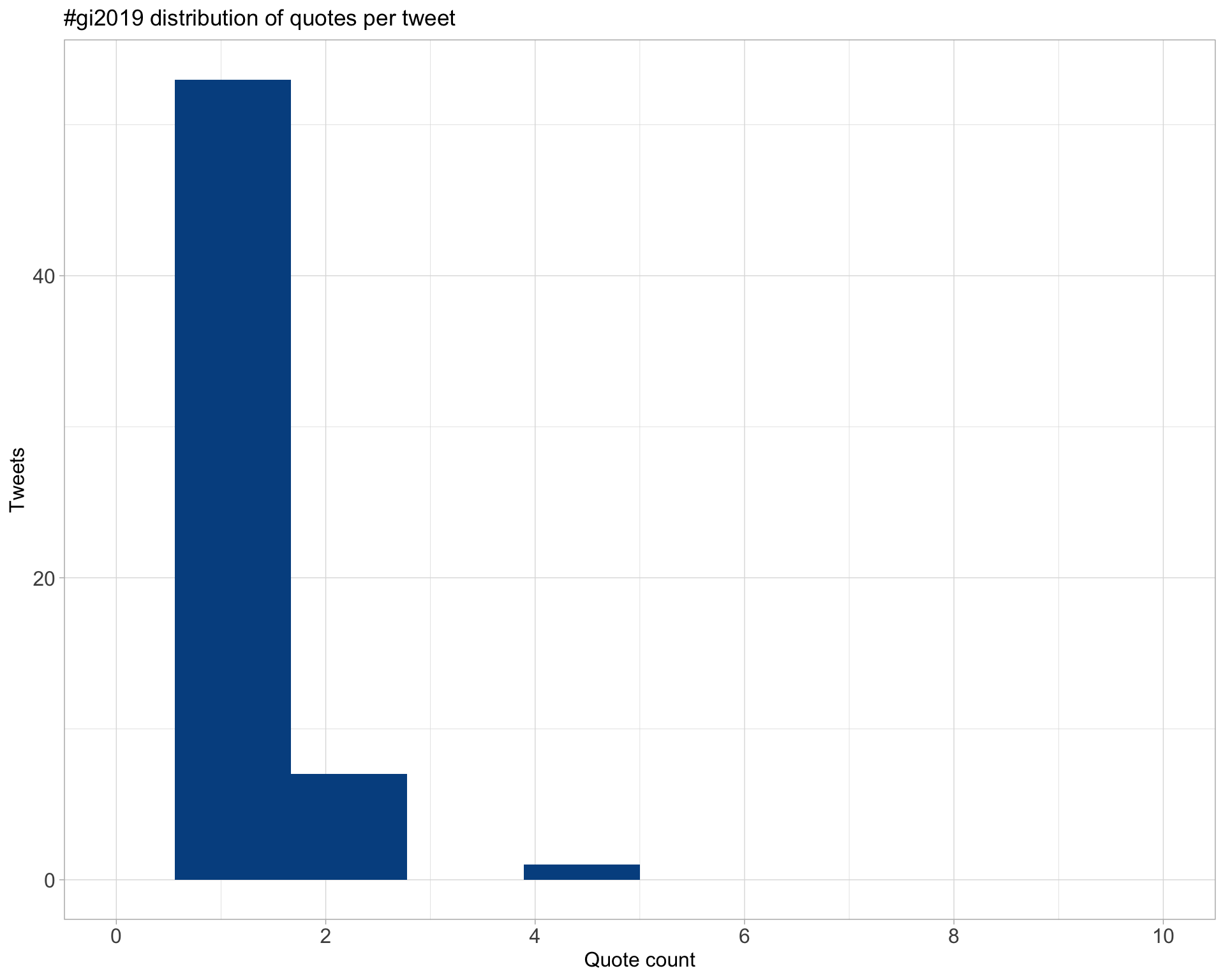
Top 10
| screen_name | text | quote_count |
|---|---|---|
| AliciaOshlack | We already have two new posters on our @F1000Research Channel for this year’s conference #Gi2019. Check them out at your leisure https://t.co/Qi3BJYQCs5 | 4 |
| AliciaOshlack | We already have 5 posters up on the website for you to look at before the poster session #GI2019 https://t.co/xrktvzLo6G https://t.co/qSNoUoImSe | 4 |
| dsurujon | What a great idea! Here are my slides for my #gi2019 talk on Friday on how transcriptomic chaos can predict bacterial fitness https://t.co/zXVnqfAcfx https://t.co/4DBC2pID7I | 4 |
| ascendox | RT mike_schatz "RT AliciaOshlack: We already have two new posters on our F1000Research Channel for this year’s conference #Gi2019. Check them out at your leisure https://t.co/pz1UEPRaQ3"; | 4 |
| AdamJOrr | Guest tweeter in @lh3lh3 ’s #gi2019 talk https://t.co/oLWLyS6ucx | 2 |
| nephantes | Heng Li @lh3lh3: nextflow pipeline used for liftover hg38 -> hg19 ☑️ sucessfully. :) @nextflowio #gi2019 https://t.co/zdDOrcO8zo | 2 |
| AliciaOshlack | Sad that @sexchrlab wasn’t able to be here to do the opening of #gi2019 with us because of stupid flight cancellations https://t.co/DbTTm0vPsr | 2 |
| AliciaOshlack | I actually really like this photo. Who would have thought that @pathogenomenick and I only met for the first time 10 mintues before that #gi2019 https://t.co/DbTTm0vPsr | 2 |
| aphillippy | Heng is amazing. Go work for him. If he turns you down, come talk to me. I am also looking for students and postdocs at #gi2019 😆 https://t.co/uuuMq6gXLJ | 2 |
| michelebusby | #gi2019 PhDs: Please don’t take yourself out the running for this because you’ve been using Heng’s tools for the last 5 years and are intimidated! I always learn a lot when I talk to Heng (as does the whole community) and this is a great opportunity. Take your shot. https://t.co/5grj3gBP1j | 2 |
Most quoted
6 Media
Proportion
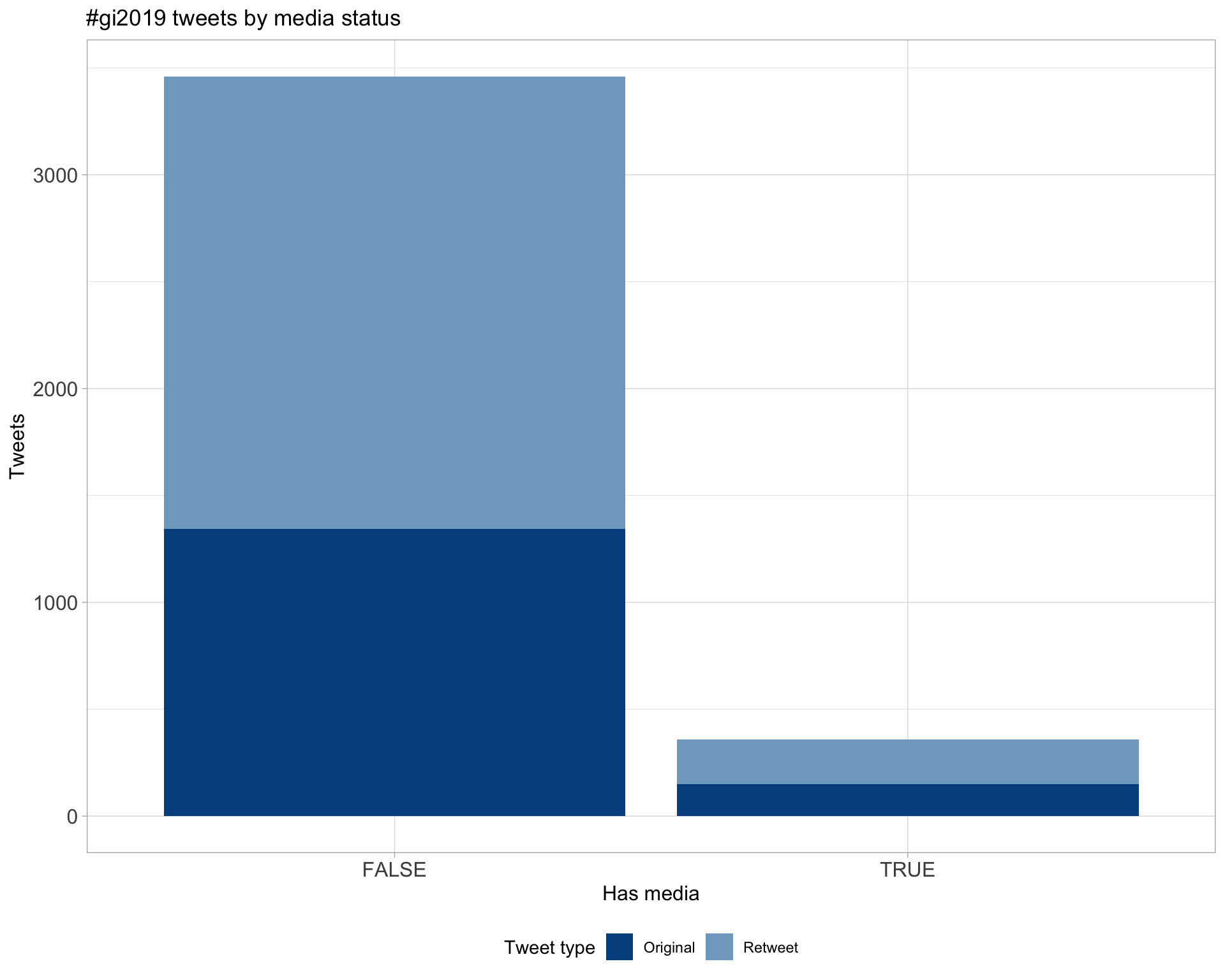
Top 10
| screen_name | text | favorite_count |
|---|---|---|
| samstudio8 | Made it to #gi2019 at @CSHL. My metagenomic assembly graph artwork is the book cover! https://t.co/T2X6EPqweC | 187 |
| ACSCevents | Save the date - Genome Informatics 2020 - 14-17 Sept at the genome campus #GI2020 #gi2019 https://t.co/EfCc74qrCR | 136 |
| infoecho | A Manhattan plot for #GI2019 https://t.co/Jpamfgex8e | 108 |
| lpachter | We’ve posted the kallisto | bustools poster presented by @sinabooeshaghi at #gi2019 on @F1000Research https://t.co/EVxAXZvGAC https://t.co/inzS4JAeUZ | 89 |
| infoecho | Real bioinformatics “file parsing”!!#gi2019 https://t.co/4vll3qI61u | 74 |
| cshlmeetings | As #gi2019 wraps up, we’d like to thank meeting sponsor @genome_gov; co-orgs @pathogenomenick, @AliciaOshlack and @sexchrlab; and those who live-tweeted during the entire meeting (especially during the lightning talk session)! See everyone at #gi2021! https://t.co/awkC8GZHTW https://t.co/RAEBm3zce3 | 70 |
| aphillippy | @AliciaOshlack and @pathogenomenick kicking off #gi2019. So many legendary bioinformagicians in attendance. Love this meeting! https://t.co/BIMjUNC1E2 | 65 |
| yoson | Guess which session this was (poll below) #gi2019 https://t.co/UBQJZdFLyn | 57 |
| AliciaOshlack | Chalkboard update for Genome Informatics meeting 2020 and 2021. Put it in your diaries. #gi2019 https://t.co/3fwbfGaVsx | 47 |
| iddux | @samstudio8 at #gi2019 on the difference between CPU and GPU tasks. “CPU is like two postdocs, they can do complex work but are easily distracted and need to do other things too. GPU is like an army of babies that can do just one simple thing, but can do it quickly and forever” https://t.co/rjQMzm7Tv3 | 45 |
6.1 Most liked image

7 Tweet text
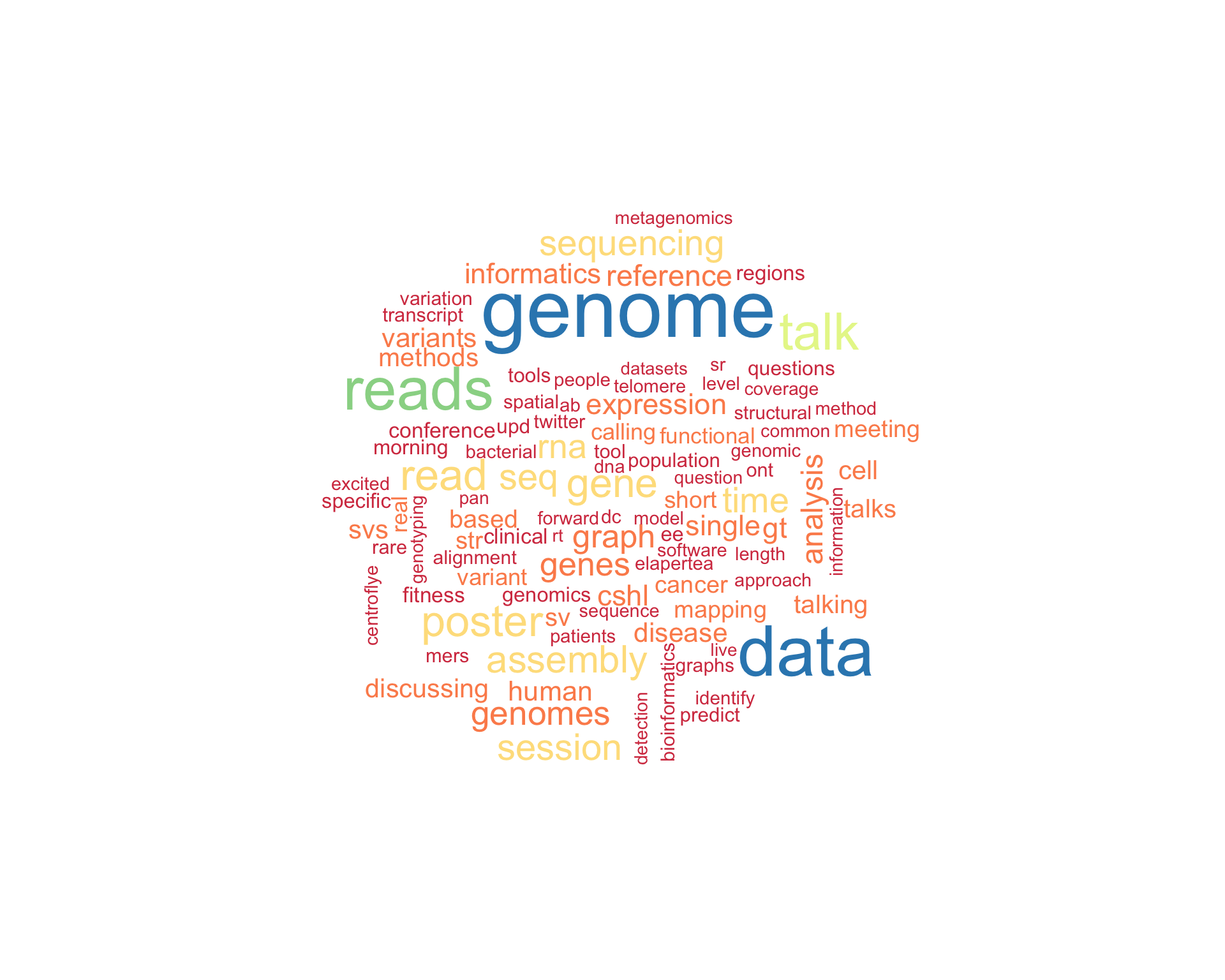
7.1 Word cloud
The top 100 words used 3 or more times.
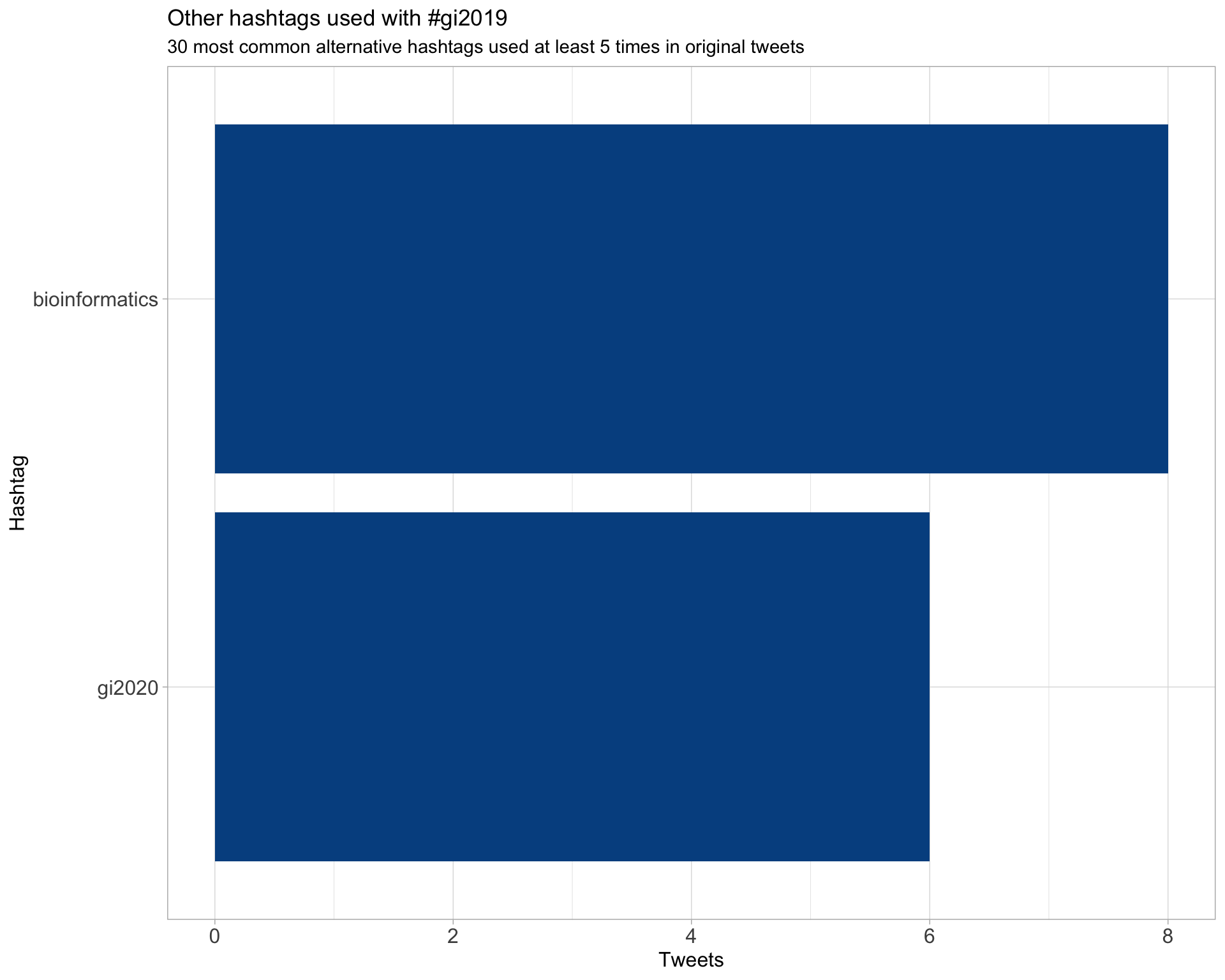
7.2 Hashtags
Other hashtags used 5 or more times.
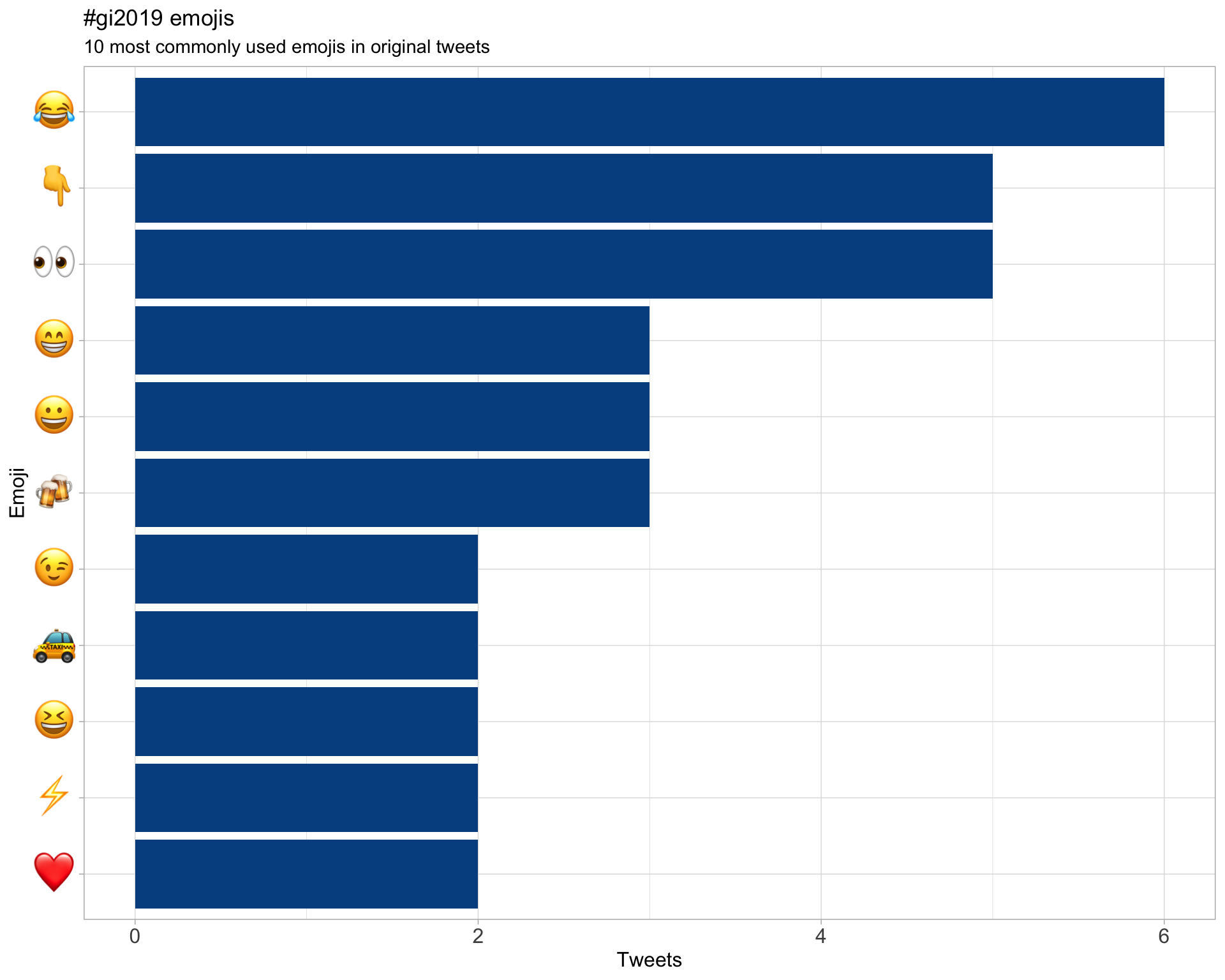
7.3 Emojis
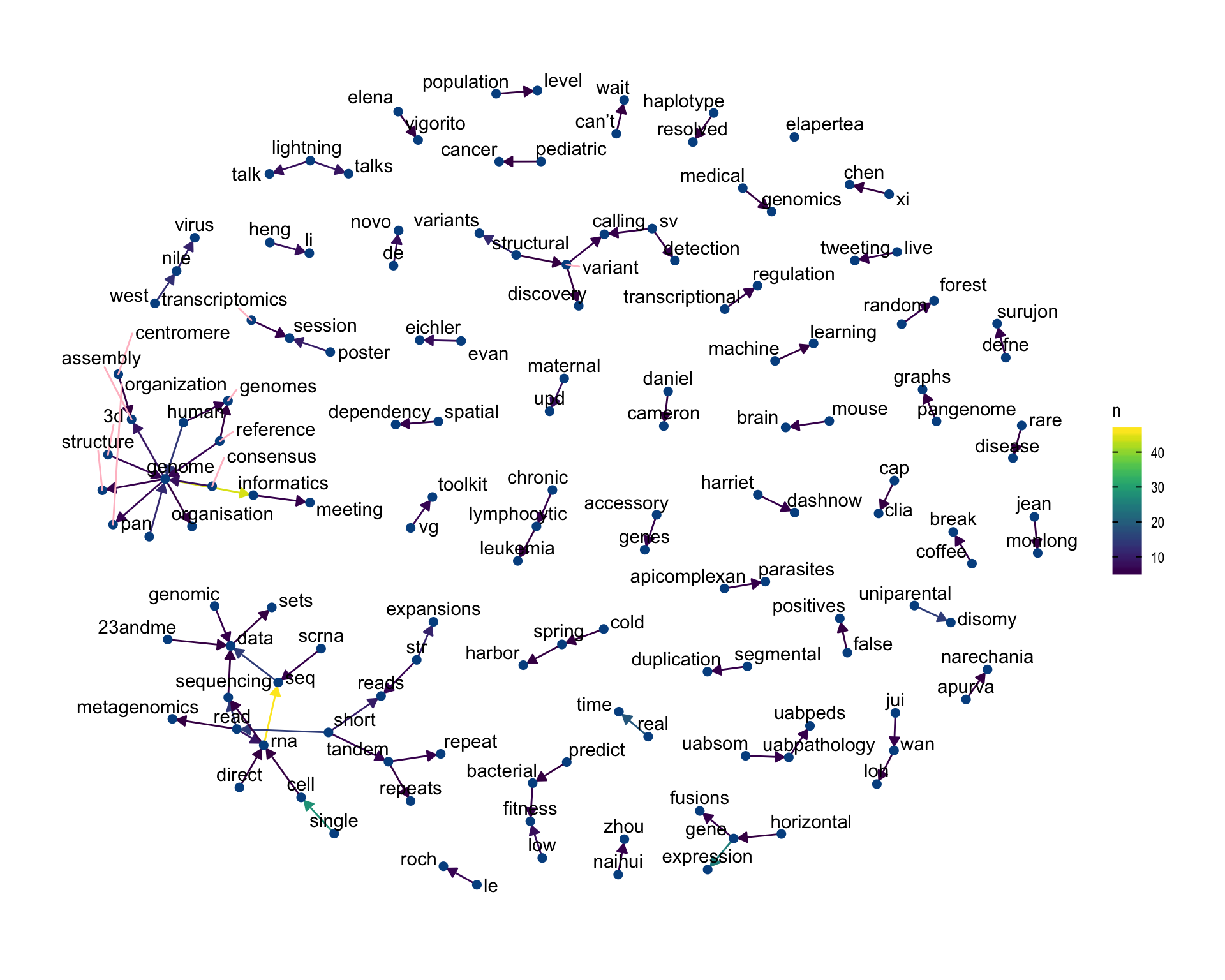
7.4 Bigram graph
Words that were tweeted next to each other at least 5 times.
7.5 Topic modelling
Top 10 words associated with 9 topics identified by LDA.
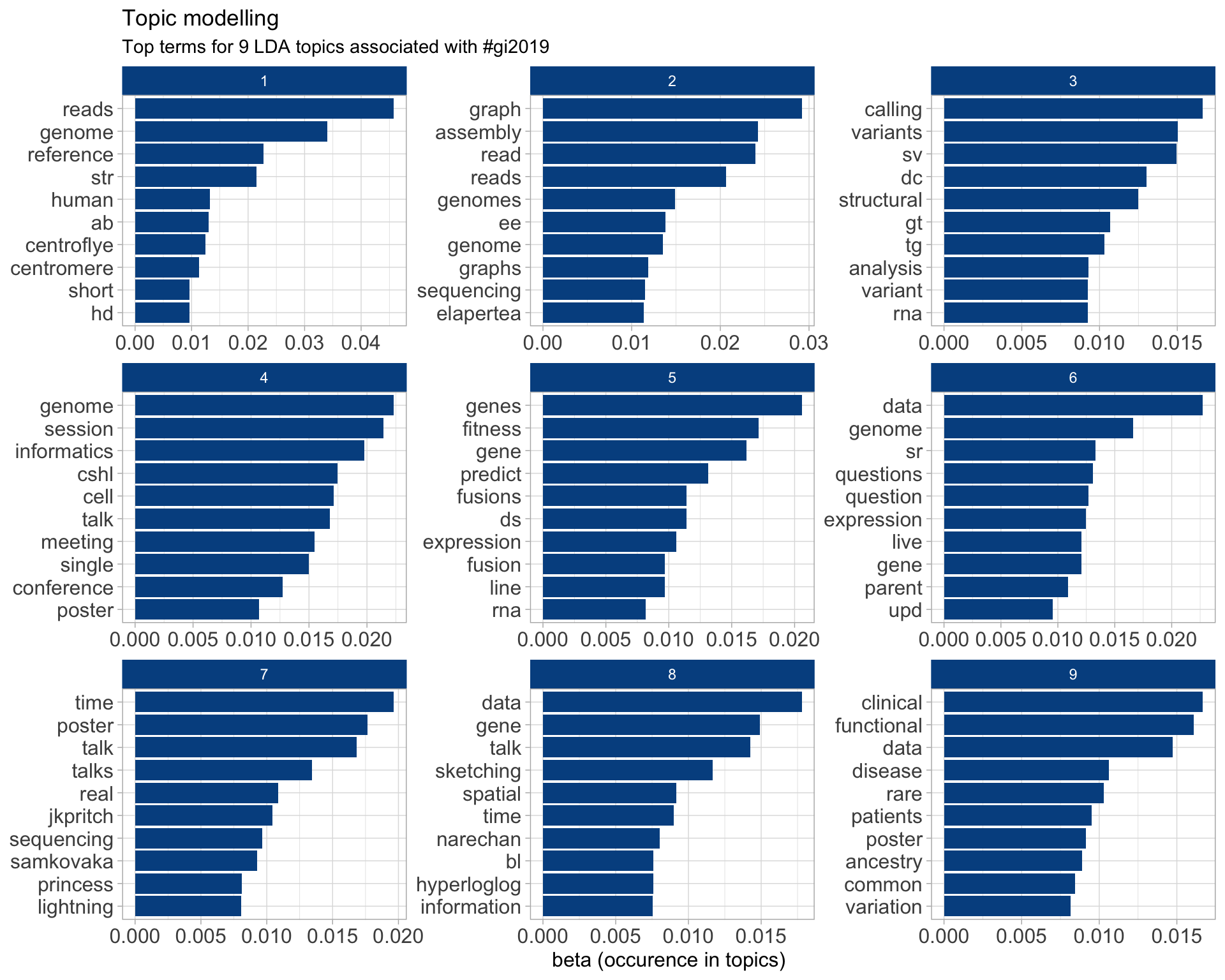
7.5.1 Representative tweets
Most representative tweets for each topic
Topic 1
| screen_name | text | gamma |
|---|---|---|
| Refasho | The fitting shoes #PixarSoul #gi2019 #WIZONELOVEIZONE #ThankfulThursday #BWFWarren #Obsession #ODMUnleashesGoons #JusticeForAsim #1026大切なユタの日 #10yearsofdanandphil #10월에_태어난_병아리_윈윈 #nyc #ny #344 #bloomfieldav #uppermontclair #montclair #newark #kearny #edison #byc https://t.co/efM3QsnsQH | 0.9905997 |
| Magdoll | Bzikadze: centroFlye for centromere assembly - identify centromeric reads, classify as prefix/internal/suffix, find rare/unique k-mers –> represent reads as k-mer clouds –> build contigs –> polish. Rub: need to deal w errors #gi2019 | 0.9899113 |
| pmelsted | STR: several methods for reference STR (all using puns including the letters STR). Canvas (ataxia) disease, reference genome has alu element and AAAAG_n motif, disease elements have AAGGG_n motif, reference based methods fail to detect these #gi2019 | 0.9895278 |
| pmelsted | SK: can get 4x enrichment of a fungal genome within a bacterial community. Human gene enrichment, 28 genes from breast cancer panel. Mapping to a 3.5M reference, eject reads after 3sec. 2.8x enrichment. Showing fresh results obtained on Monday #gi2019 | 0.9895278 |
| pmelsted | SK: ONT real time, read-until can monitor the sequence, up to 512 channels at 450bp/sec. UNCALLED maps 10kbp/sec per thread, 75% of reads classified within 1 sec, [aside: ONT can eject reads from pores, saving on sequencing]. #gi2019 | 0.9886662 |
| lizworthey | KR: multiple global genomic measures being considered; telomere length, genome complexity, coding mutational burden, mutational signatures describing factors that have impacted genome e.g. smoking/age #gi2019 | 0.9876502 |
| pathogenomenick | Andrey Bzikadze talking about centromere assembly with centroFlye: bigging up @khmiga and @aphillippy’s burgeoning T2T Consortium data which has huge haul of ultra-long reads and complementary technologies for human haploid genome: https://t.co/bNU6QpvEfI #gi2019 | 0.9876502 |
| ascendox | RT mike_schatz “RT Magdoll: Aganezov: how to retrace cancer rearrangement history? formulate as minimal eulerian decomposition problem. to deal with non-unique solutions, form as consistent contig covering problem (<–this is new to me). #gi2019 prep… https://t.co/tKWOjW7ITd” | 0.9870707 |
| pmelsted | AB: length of centromere of X is ~2.8Mbp, consists of repeats of 171bp repeats, in 12 units that are DXZ1. Centromeres are 3% of reference, play a critical role in chrom segregation, variations are linked to cancer and infertility #gi2019 | 0.9870707 |
| bnlasse |
#gi2019 @AndreyBzikadze: centroFlye-Assembling centromeres w/ long error-prone reads Telomere-to-tel consortium: https://t.co/HdmPfXmTTk https://t.co/Sg82pHMR3q centroFlye: ID unique k-mers in centromere, use for centro. assembly https://t.co/X6KKGbvJmv https://t.co/0luMvnOzYm |
0.9864341 |
Topic 2
| screen_name | text | gamma |
|---|---|---|
| pmelsted | HL: Incremental graph construction. Invariant property: linear path (original genome) and acyclic. Seq to graph alignment: Find seeds in segments, use linear chaining ignoring topology. Graph chaining, use graph topology to line up chains. Search best path with heuristics #gi2019 | 0.9905997 |
| bnlasse |
#gi2019 Heng Li @lh3lh3 The construct & utility of reference pan-genome graphs Plan: start w/ GRCh38, incrementally add other genomes to construct graph, blacklist & decoy seqs for linear tools, updates preserve coordinate sys https://t.co/B6XVbWQX50 https://t.co/NcTsAbyPyg |
0.9899113 |
| pmelsted | HL: VCF, hard to represent small variants on long insertions wrt reference. GFA format, two components. S-line segments that are sequences, L-line: links between segments. Most popular assembly format. GFA coordinates are unstable, can split segment and change coordintes #gi2019 | 0.9899113 |
| bnlasse |
#gi2019 Shilpa Garg: Efficient chr-scale haplotype-resolved assembly of human individuals method leverages long accurate reads+long-range conformation data for individuals to generate chr-scale phased assembly w/in a day 👀👇 https://t.co/4M17NhWId8 https://t.co/XhXlNj9TBl |
0.9895278 |
| bnlasse |
#gi2019 M Pertea, Efficient & robust transcriptome reconstruction from long-read RNA-seq alignments Based on StringTie, short read transcriptome assembler: https://t.co/Kjg3ng0GPt StringTie2, long read sln, overcome error rate and lower throughput 👀👇 https://t.co/00KzRLeRnH |
0.9891140 |
| pmelsted | Heng Li, @lh3lh3: Pangenomes, construction and utility. Styles of pangenome graphs: assembly style graph (large messy), linear graph (string of bubbles). Linearity no 2 bases on a haplotype can be mapped to the same graph position. VCF is linear #gi2019 | 0.9886662 |
| pmelsted | HL: sad stories about Gnomad taking 6 years to move to Grch38 and someone running liftover to hg19 from Gnomad v3. New restricted format rGFA. Can incrementally add genomes to construct a graph. Updates preserve coordinate system #gi2019 | 0.9886662 |
| kasukawa |
Utilization of consensus genome, in which minor alleles are replaced to major ones. Interestingly, pan-human consensus genome is adequate to reduce error rates. Even if using consensus genomes specific to subpopulations, the error rates were not reduced. #gi2019 |
0.9881800 |
| pmelsted | EE: Phased assembly, uses linked reads to phase, hifi reads to construct contigs. 81% of hifi reads can be assigned to a haplotype. Regions that were problematic for canu and peregrine assemblers, about 250, colocalized with segmental duplications. #gi2019 | 0.9876502 |
| lizworthey | MK: discussing cow rumen metagenome 11.4 Gb, N50=11Kb. assembly 268 Mr, N50=230Kb. 44 contigs> 1Mb After assemble using tools found 649 bubbles, 96 superbubbles, 210 shared bubbles #gi2019 | 0.9876502 |
Topic 3
| screen_name | text | gamma |
|---|---|---|
| pmelsted | TG: Filtering FPs using dual strand filter, nanopolish has highest sensitivity at high coverage. Phased variants in TP53 region using WhatsHap. Phasing of normal vs tumor tissue revealed cancer specific snps and a copy number difference between alleles in tumor #gi2019 | 0.9895278 |
| pmelsted | TG: High coverage of targeted regions means you can call SNPs better (except for homopolymer sites). Comp of tools, samtools, clair, medaka and nanopolish. 0.98 sensitivity for nanopolish and samtools at 200x cov (still a number of FPs), FPs often from a single strand #gi2019 | 0.9891140 |
| pmelsted | Daniel Cameron on structural variation and CNV from cancer samples. Fully automated oncology reports, no possibility of manual intervention. Wrote 3 software for SV calling (GRIDSS), CNV calling (PURPLE) and interpretation (LINX) #gi2019 | 0.9881800 |
| chrisamiller |
DC: How can we make good, clinic-ready structural variant calls from tumor patients?
|
0.9881800 |
|
YiXing77 |
My student Yang Pan @ypnngaa will present his work on PEGASAS, pathway guided analysis of alternative splicing, at the upcoming Penn #RNA club next Tuesday! He also has a poster about PEGASAS at CSHL #gi2019 this week. #IOTN #Moonshot https://t.co/jzIJSD6wWU |
0.9876502 |
|
Repealist_ |
Commuting to @CSHL #gi2019 meet and stopped off at Sutphin Blvd waiting for a train Have already talked to three Bangla families and one Korean gal And the morning fishmarkets are truly the view of dreams One for you @MichaelsCoDub https://t.co/glRsqxmruN |
0.9857315 |
| lizworthey | DC: developed a set of tools to better call SVs. Wrote a new GRIDSS based tool for SVs, Purple specifically for CNV, and a tool called LINX for variant interpretation #gi2019 | 0.9857315 |
| byuhobbes | @BenLangmead alternative to the bottom k approach (aka min hashing) is k-partition approach; break up k-mer space into k partitions, take the bottom element of each partition; similar characteristics to min hashing #gi2019 | 0.9857315 |
| Magdoll | Gangavarapu: West Nile virus shown to persist through the seasons and circulate across all regions in California. Unable to link it to surviving in birds (or mosquitos? someone correct me) in winter (no seasonality). #gi2019 | 0.9849523 |
| byuhobbes | @MedhatHelmy7 @MedhatHelmy7 Benchmarked variant calling and phasing tools with GIAB data catalog; better SNV calling performance with 10x coverage from PacBio CCs vs 25x from PacBio CLR or 25x from ONT #gi2019 | 0.9849523 |
Topic 4
| screen_name | text | gamma |
|---|---|---|
| bnlasse |
#gi2019 A Balsubramani; Multi-resolution, interactive, atlas-scale integration of single-cell assays & exper. Unify datasets from diff experiments? Connect w/ anchors, impute/co-embedding, refine on subpops and repeat. Their approach=Musica [I don’t think released quite yet] |
0.9891140 |
| lpachter | Now Akshay Balsubramani talking about integrating data from single-cell RNA-seq experiments (w/ @anshulkundaje). This is an important topic. Full disclosure: we’ve been partial to @_romain_lopez_’ et al.’s scVI in my lab but constant exciting new ideas in this area. #gi2019 | 0.9864341 |
| Magdoll | Balsubramani: unifying single cell RNA-seq experiments. steps: anchor (find cells of similar cell types) –> impute features –> refine co-embedding. repeat. #gi2019 | 0.9864341 |
| byuhobbes | @Magdoll @Magdoll Suggests that single-cell platforms produce shorter transcripts than bulk full-length cDNA; using proximity to CAGE peaks as orthogonal evidence of 5’ completeness; polyadenylation signals for 3’ completeness #gi2019 | 0.9857315 |
| lpachter | I highly recommend “How to read (single-cell RNA-seq) PCA plots” (by @vallens): https://t.co/Co3A47SZTR #gi2019 He notes that “A particular danger [in interpreting T-shapes] is that it is tempting to interpret this as a bifurcation in the data.” (it almost never is). #gi2019 | 0.9857315 |
| lpachter | A special privilege of attending a @CSHL meeting is that one can learn about recent research via glimpses at unpublished data. But when it comes to methods talks at meetings such as #gi2019 I think there should be a public Github repo or equivalent. https://t.co/kYT0vLduvQ | 0.9857315 |
| sexchrlab |
In past years, we tried to add a place for abstract submitters to add a link to software repos/web interfaces; I think this would be great to bring back. Genome Informatics slides and posters have and can be posted here (please share!): https://t.co/aWCJRpKmnD #gi2019 |
0.9849523 |
| YiXing77 | @DNAwyman of UCI presenting the ENCODE TALON software for long read RNA-seq data analysis https://t.co/7QDZvNwxcn. Also nice to see a figure from our AJHG review https://t.co/QS52kMJaiB by @RNAEddie used in the Intro! #gi2019 https://t.co/MhKOMKNUxO | 0.9849523 |
| cshlmeetings | Meet @samstudio8 of @unibirmingham! He’s busy at #gi2019. He gave a talk, his work is featured on the cover of the meeting’s abstract book, and is meeting in person the Twitter avatars with whom he’s been messaging the past year or so! #cshlvisitor https://t.co/6HmQ9eKFCb | 0.9840830 |
| winhide | #gi2019 we are looking for a post doc who wants to work with an amazing team (I mean it!) Want to find the best drug that drives resilience against Alzheimer’s? Join us! - the NIH funded this work. Chat to Sarah Morgan at the meeting. https://t.co/0b4pyljOJl | 0.9831071 |
Topic 5
| screen_name | text | gamma |
|---|---|---|
| lizworthey | SLH: Process is RNA-seq, run data (on Dragen on AWS) through FusionMap, JAFFA, STARfusion, FusionCatcher, MapSplice, and TopHat fusion with overlap analysis, hierarchical prioritization, knowledge based filtering, and then CAP-CLIA validation of clinically relevant vars #gi2019 | 0.9902676 |
| bnlasse |
#gi2019 S LaHaye: Utilization of ensemble approach for ID of driver fusions in peds cancer Ensemble approach reduces false +s 6 tools (FusionMap, JAFFA, STARfusion, FusionCatcher, MapSplice, TopHat Fusion) DRAGEN to speedup [seems to ID 98% in test] Seraseq as truth |
0.9899113 |
| chrisamiller | SL: Using a ensemble approach of 6 gene-fusion callers to identify fusions in ped cancer. Overlap (by at least 2 callers), prioritize based on evidence, and compare to a knowledge base. Final step is CAP/CLIA RT-PCR or sanger validation #gi2019 | 0.9895278 |
| bnlasse |
#gi2019 Xi Chen, ‘Tissue-specific enhancer functional networks for associating distal regulatory regions to disease’ How interpret/predict fxn of enhancers/disease-associated enhancers? Bayesian inference: https://t.co/JjuT1Ar8pq (try it!) autism e.g.: https://t.co/TlWmLl7CXY |
0.9886662 |
| lpachter | @sinabooeshaghi @GoogleColab @jspacker @jisaacmurray @JunhyongKim @sinabooeshaghi: kallisto | bustools runs up to 100 times faster than Cell Ranger, 25 times faster than Alevin and 4 times faster than STARsolo https://t.co/GGveUbyaUu Many features and capabilities not available with other tools. See poster this afternoon at #gi2019. | 0.9876502 |
| chrisamiller |
LINE1 activity correlates with poor immunotherapy response. Suggests that LINE1 high tumors are cloaked in a way that’s sidesteps activation with std imm. therapy (if they weren’t cloaked, they’d already be gone, since high LINE1 triggers response in normal cells!) #gi2019 |
0.9870707 |
| lizworthey | gloria integration of ALzheimers disease genetics and myeloid cell genomics to revel novel risk mechanisms HiC, epigenomics, GWAS hits to identify causal genes and loci and validated in microglia #gi2019 | 0.9870707 |
| lizworthey | anoushka joglekar on enhanced interpretation of single-cell isoform RNA sequencing to reveal distinct programs of alternative splicing in the developing mouse brain correlated to RNA splicing machinery #gi2019 | 0.9870707 |
| Magdoll | .@dsurujon : gene expression is more chaotic in low fitness bacteria, this means using entropy (measuring chaos in gene expression) can predict fitness. but need to model for co-expressed genes that break the independence assumption. #gi2019 | 0.9864341 |
| lizworthey | Katherin Woolley-Allen asking what is the weakest level off transcriptional factor binding that can exert an effect; evolution has gone further towards lower and lower affinity binding sites acting in combination #gi2019 | 0.9849523 |
Topic 6
| screen_name | text | gamma |
|---|---|---|
| byuhobbes | @s_ramach @s_ramach Question: how are ROHs distributed in the genome? Parent-child data in 23andMe has revealed extremely long IBD segments in putatively normal individuals; reflection of uniparental disomy, where both haplotypes inherited from a single parent #gi2019 | 0.9876502 |
| bnlasse |
#gi2019 Karine Le Roch, ‘Comparative 3D genome organization in Apicomplexan parasites’ Overall, work supports link between genome organization and gene expression in more virulent pathogens learn more from their recent pubs: https://t.co/0bG5uj8tvD https://t.co/Fc3Ar3U8re |
0.9876502 |
| lizworthey | SA: using a combination of 30x 10X, 40x ONT, and 40x PacBio and aligning with Long ranger and NGMLR. calling with lumpy, manta, SvABA, GROCSV, NAIBR, LongRanger, sniffles, PBSV and combined using survivor methods #gi2019 | 0.9876502 |
| GenomeBiology | SR: has been looking at runs of homozygosity (ROH) in 23andMe data. Parent-child data reveals extremely long shared segments eg child has whole chromosome with no sequence shared with father - called uniparental disomy #gi2019 | 0.9870707 |
| CalvinH43357785 |
Uncanny Attraction #astroworldfestival #AUSvPAK #Bigil #BREAKING #crush #ChangeForWonho #ChaseYoung #DeathStrading #DoctorSleepMovie #EricCiaramella #EpsteinSuicideCoverUp #EXOonearewe #Ethiopia #FlashbackFriday #FursuitFriday #gi2019 #GymJordan #G_I_DLE https://t.co/FZVvn45dCp |
0.9864341 |
| TwilightForce1 |
sPaCe oVeRtAkEn #astroworldfestival #AUSvPAK #Bigil #BREAKING #crush #ChangeForWonho #ChaseYoung #DeathStrading #DoctorSleepMovie #EricCiaramella #EpsteinSuicideCoverUp #EXOonearewe #Ethiopia #FlashbackFriday #FursuitFriday #gi2019 #GymJordan #G_I_DLE https://t.co/HAxVeeEsFk |
0.9864341 |
| Magdoll | Nakka: runs of homozygosity (ROH) are associated w divergence from Africa. Diff classes of ROH have diff mean/variance in population. How to study ROH distribution within a genome? @23andMe parent-child data! #gi2019 | 0.9864341 |
| kasukawa |
quantifying isoform expression in scRNA-seq data with STARsolo-Quant #gi2019 Quantifying each isoform based on the distribution function (by Maximum likelihood estimation) for distance to TTS and UMI barcoding. |
0.9864341 |
| lpachter | Interesting mention of kernel Canonical Correlation Analysis (KCCA) at #gi2019 by @KarineLeRoch1 for coupling genome organization data with gene expression data. Nonlinear CCA is complex and still not widely used in genomics. Related: https://t.co/ux3wgq7XpQ | 0.9864341 |
| Magdoll | Nakka: mining 23&me parent-child data revealed cases of uniparental disomy (UPD) where both chromosomes of a parent is preserved with no copy from the other parent. This occurs due to non-disjunction during meiosis O__O #gi2019 | 0.9857315 |
Topic 7
| screen_name | text | gamma |
|---|---|---|
| lizworthey | KG: main goal of consortia is to provide real-time high definition reconstructions of the micro- (e.g., city/county) and macro-level (e.g., state/country) spread and evolution of WNV by sequencing from infected birds, mosquitoes, and patients #gi2019 | 0.9891140 |
| Magdoll | Dilthey: Applies MetMaps to PacBio HMP & ONT Zymo mock community achieves high read assignment on both. Caveat: reduced recall due to min RL 1kb; HMW DNA required. #gi2019 Paper here: https://t.co/KxePwF8jwM | 0.9881800 |
| byuhobbes | @jkpritch @jkpritch Recent metaanalysis of schizophrenia GWAS studies found 108 genome-wide significant loci; most hits are difficult to interpret, and only explain 10% of the observed heritability (“missing heritability” problem); enormous number of small effects #gi2019 | 0.9876502 |
| FadelBerkadar |
A few words about novoSplice poster for the Genome Informatics meeting at CSHL #gi2019
|
0.9870707 |
| BBFCreativeEnt | It must suck to support a racist president. This quote will bite you in the ass when you are up for reelection….It must suck to loose BIG to a democrat…and have to work in the private sector!#ThursdayMotivation #redcup #gi2019 #WashingtonPost #NYTimes #art #washingtinTimes https://t.co/aK9vZlnawo | 0.9864341 |
| Magdoll | Mahmoud: PRICNESS: mapping (NGMLR/minimap2), SNV (Clair), SV (Sniffles), Phasing (WhatsHap, Princess-subtools), optionally can use parental SNPs and methylation (nano polish) . Also stats. #gi2019 | 0.9864341 |
| ascendox | RT mike_schatz “RT StevenSalzberg1: SamKovaka talking about”read until" sequencing on nanopore sequencers. Still experimental, but very cool that the sequencer can recognize DNA species in real time, and stop sequencing a read by reversing the current #gi2019" | 0.9864341 |
| lpachter | at #gi2019 RP is suggesting that transcriptome alignment (substrate for RSEM, eXpress, kallisto..) is not as good as genome alignment. A comparison was performed by @alexlachmann et al. in https://t.co/Bg5QhHA4EY. They conclude “the quality of the predictions is almost identical” https://t.co/M8XM0eKAer | 0.9864341 |
| David_McGaughey | Made it to #gi2019. First time staying at cshl Banbury campus. If you do scRNA stuffs make time to bother me about me 750k+, 14 study ocular integration project while you wait to talk to @pmelsted at poster session 1 (105). https://t.co/D87VAc39T9 | 0.9857315 |
| lizworthey | philip kleinert; CADD-SV tool to score the effect of SVs - whether they have strong effect on health by training to differentiate between benign and pathogenic. Use evolutionary motivated approach #gi2019 | 0.9849523 |
Topic 8
| screen_name | text | gamma |
|---|---|---|
| bnlasse |
#gi2019 Naihui Zhou: Exploring 3D spatial dependency of gene expr using Markov random fields hypothesis: expr is spatially dependent developed PHiMRF https://t.co/YQjn6CRXGO prob model for RNA-seq data accounting for gene locals in 3D genome (spatial dependency for count data) |
0.9914725 |
| bnlasse |
#gi2019 Sam Kovaka: Rapidly mapping raw @nanopore signal w/ UNCALLED to enable real-time targeted seq ReadUntil seq bg: https://t.co/L5obZVjusq UNCALLED: Maps raw nanopore signals from fast5 files to large DNA refs (preprint soon!) https://t.co/gyahq5GDBd |
0.9895278 |
| Repealist_ |
Incidental window seat right near the front, space to get work done, and it starts raining just as I’m leaving Dublin? All the promising makings of a great journey to #gi2019 and #SUNY. Slán a chairde 🇮🇪 tá sí ag dul go Nua-Eabhrac 🇺🇸! https://t.co/GTcyz95iNn |
0.9881800 |
| pmelsted | BL: Hyperlog (HLL) can process array of bits using SIMD values. Uses a bit more space by using a different base 1.19, fills up 8-bits rather than 6-bits with base 2. HLL performs better with lopsided data (large ratio of sizes between two sets) #gi2019 | 0.9881800 |
| lpachter | I agree we should not hold scientific meetings on weekends. I also think we should not hold scientific meetings on weekdays. Scientists with kids face enormous logistical challenges on weekdays (kid pickup/dropoff, school events, activities, etc.) #gi2019 https://t.co/ifuJZuV3ci | 0.9876502 |
| chrisamiller | FN: answering why ancient LINEs so frequently show up in RNAseq. To make a long story short, seq similarity causes mismapping, and their tool TeXP can correct for this. Shows that basically all autonomous transcription is from recent families, as expected #gi2019 | 0.9864341 |
| bnlasse |
#gi2019 Ben Langmead: Genomic sketching w/ HyperLogLog Sketching=how to sift&summarize huge datasets so can answer similarity ?s later Dashing w/ HyperLogLog https://t.co/5MWtP6xfPD https://t.co/byoPzN12XA https://t.co/GSykouIW4c [review of interest: https://t.co/OO5AklzeAz] |
0.9857315 |
| GrameneDatabase | If you missed @ajo2995 Andrew Olson’s delightful talk on Crop PanGenome Sites at #gi2019, look for us at the @cshlmeetings Wine and Cheese to savor a TE Cabernet and avoid being haunted by the ghost of Barbara McClintock https://t.co/eM88kVKzXv | 0.9857315 |
| Magdoll | plot twist: Narechania, just like everyone else, is ditching base-alignments and going for k-mers. Alright, k-mer based ortholog matrix to infer HGT. But too many k-mers, so gotta compress them without info loss. #gi2019 | 0.9857315 |
| hdashnow | Yesterday I got my official PhD passed email while I was on stage at #gi2019 talking about work I’ve done since submitting my PhD in June. That is considered a relatively fast turn-around for an Australian PhD assessment! Thanks for being an awesome PhD supervisor @AliciaOshlack | 0.9849523 |
Topic 9
| screen_name | text | gamma |
|---|---|---|
| Magdoll | (not related to #gi2019 but another TV idea - patient is admitted to Nationwide Childrens Hospital w rare disease and - panning to dramatic gradient boosted decision tree - reveals the patient has some ancestry from exotic land coinciding w mother’s younger traveling years) | 0.9886662 |
| bnlasse | @lizworthey @UABSOM @UABPathology @uabpeds #gi2019 @lizworthey showing examples of software developed by her group for use by non-informatics experts and cases evaluated by her team’s secondary analysis pipeline including a ‘B-allele’ tool to look for chimerism/mosaicism missed by traditional pipelines | 0.9870707 |
| kasukawa |
Plant genomes. Highly variable in the maize genome. Gramene is a curated, open-source, integrated data resource for comparative functional genomics in crops and model plant species. https://t.co/8TFAvyA56a #gi2019 |
0.9864341 |
| GenomeBiology | KR: looking at chronic lymphocytic leukemia. More prevalent in men than women. Patients can survive for years without treatment. 8-12% of patients have TP53 mutation, which corresponds with lower survival. Over 50% of patients with refractory CLL have no known marker #gi2019 | 0.9857315 |
| Ailith_Ewing | Anyone interested in chromothripsis in high grade serous ovarian cancer? Check out Stuart Brown’s poster this afternoon! And chat to Stuart, a PhD student with @DrColinSemple, Charlie Gourley and myself, to hear all about it! #gi2019 | 0.9849523 |
| bnlasse | @lizworthey @UABSOM @UABPathology @uabpeds #gi2019 sequencing approaches have been great, also seeing advances in software engineering/genome informatics/comp bio allowing us to identify other important variation (e.g., mobile insertions, etc.) @lizworthey | 0.9849523 |
| byuhobbes | MM: SURFDAWave also trained a model to discriminate between neutral, sweep, and adaptive introgression; classification performance increased by supplementing 1D stats with 2D stats #gi2019 | 0.9840830 |
| lizworthey | AR: In house dataset contained pedigree data allowing access to confirmed parental ancestries so were able to determine if methods accurately predicted the composition in such individuals #gi2019 | 0.9840830 |
| lizworthey | Jiao Sun discussing computational methods to explore the role of post-transcriptional regulation in cancer; including integrative model for alternative polyadenylation #gi2019 | 0.9831071 |
| bnlasse | @lizworthey @UABSOM @UABPathology @uabpeds #gi2019 @lizworthey see another tool here: https://t.co/T7tCfipNOu; also discussing strategies they are developing for examining modifier variants that might explain differences in disease presentation and outcomes | 0.9831071 |
8 Links
Links to GitHub, GitLab, BitBucket, Bioconductor or CRAN mentioned in Tweets.
| Name | Tweets | Retweets | Type | Link |
|---|---|---|---|---|
| lancet | 2 | 13 | GitHub | nygenome/lancet |
| gridss-purple-linx | 3 | 9 | GitHub | hartwigmedical/gridss-purple-linx |
| gridss | 2 | 7 | GitHub | papenfusslab/gridss |
| STRling | 7 | 1 | GitHub | quinlan-lab/strling |
| dashing | 6 | 2 | GitHub | dnbaker/dashing |
| UNCALLED | 6 | 2 | GitHub | skovaka/uncalled |
| princess | 5 | 2 | GitHub | mehelmy/princess |
| stringtie | 1 | 6 | GitHub | gpertea/stringtie |
| Palantir | 2 | 4 | GitHub | dpeerlab/palantir |
| RCK | 2 | 4 | GitHub | aganezov/rck |
| mmseqs2 | 1 | 5 | GitHub | soedinglab/mmseqs2 |
| centroFlye | 5 | 0 | GitHub | seryrzu/centroflye |
| reticulatus | 2 | 3 | GitHub | samstudio8/reticulatus |
| MetaMaps | 3 | 0 | GitHub | diltheylab/metamaps |
| sketch | 3 | 0 | GitHub | dnbaker/sketch |
| sveval | 3 | 0 | GitHub | jmonlong/sveval |
| WHdenovo | 3 | 0 | GitHub | shilpagarg/whdenovo |
| assembly | 2 | 1 | GitHub | hsnguyen/assembly |
| minigraph | 2 | 1 | GitHub | lh3/minigraph |
| PhiMRF | 2 | 1 | GitHub | ashleyzhou972/phimrf |
| pyranges | 1 | 2 | GitHub | biocore-ntnu/pyranges |
| STRling | 1 | 2 | GitHub | quinlan-lab/strling |
| contig-covering | 2 | 0 | GitHub | izban/contig-covering |
| vg | 2 | 0 | GitHub | vgteam/vg |
| All-FIT | 1 | 1 | GitHub | khiabanianlab/all-fit |
| bustools | 1 | 1 | GitHub | bustools/bustools |
| Flye | 1 | 1 | GitHub | fenderglass/flye |
| kallisto | 1 | 1 | GitHub | pachterlab/kallisto |
| kb_python | 1 | 1 | GitHub | pachterlab/kb_python |
| RCK | 1 | 1 | GitHub | aganezov/rck |
| readfq | 1 | 1 | GitHub | lh3/readfq |
| scVI | 1 | 1 | GitHub | yoseflab/scvi |
| conterminator | 1 | 0 | GitHub | martin-steinegger/conterminator |
| dashing | 1 | 0 | GitHub | dnbaker/dashing |
| Fl | 1 | 0 | GitHub | fenderglass/fl |
| Flye | 1 | 0 | GitHub | fenderglass/flye |
| gfatools | 1 | 0 | GitHub | lh3/gfatools/blob/master/doc/rgfa.md |
| GTEx_Brains | 1 | 0 | GitHub | sexchmlab/gtex_brains |
| LayeredGraph | 1 | 0 | GitHub | hudsonalpha/layeredgraph |
| Met | 1 | 0 | GitHub | diltheylab/met |
| minigraph | 1 | 0 | GitHub | lh3/minigraph |
| nanopore-methylation-utilities | 1 | 0 | GitHub | isaclee/nanopore-methylation-utilities |
| npBarcode | 1 | 0 | GitHub | hsnguyen/npbarcode |
| npScarf | 1 | 0 | GitHub | mdcao/npscarf |
| parag | 1 | 0 | GitHub | illumina/parag |
| phimrf | 1 | 0 | GitHub | ashleyzhou972/phimrf |
| plass | 1 | 0 | GitHub | soedinglab/plass |
| prince | 1 | 0 | GitHub | mehelmy/prince |
| RefineInsertions | 1 | 0 | GitHub | mkirsche/refineinsertions |
| ret | 1 | 0 | GitHub | samstudio8/ret |
| SDA | 1 | 0 | GitHub | mvollger/sda |
| ShinyOmics | 1 | 0 | GitHub | dsurujon/shinyomics |
| sketch | 1 | 0 | GitHub | dnbaker/sketch |
| sqanti2 | 1 | 0 | GitHub | magdoll/sqanti2 |
| sv-genotyping-paper | 1 | 0 | GitHub | vgteam/sv-genotyping-paper |
| sveval | 1 | 0 | GitHub | jmonlong/sveval |
| TALON | 1 | 0 | GitHub | dewyman/talon |
| TALON | 1 | 0 | GitHub | dewyman/talon |
| TOBIAS | 1 | 0 | GitHub | loosolab/tobias/ |
| TranscriptClean | 1 | 0 | GitHub | dewyman/transcriptclean |
| TranscriptClean | 1 | 0 | GitHub | dewyman/transcriptclean |
| UNCALL | 1 | 0 | GitHub | skovaka/uncall |
| VarSight | 1 | 0 | GitHub | hudsonalpha/varsight |
| vg | 1 | 0 | GitHub | vgteam/vg |
| WHd | 1 | 0 | GitHub | shilpagarg/whd |
| wnv-phylogeography | 1 | 0 | GitHub | andersen-lab/wnv-phylogeography |
| XPRESSpipe | 1 | 0 | GitHub | xpressyourself/xpresspipe |
Session info
## ─ Session info ───────────────────────────────────────────────────────────────
## setting value
## version R version 4.0.0 (2020-04-24)
## os macOS Catalina 10.15.6
## system x86_64, darwin17.0
## ui X11
## language (EN)
## collate en_US.UTF-8
## ctype en_US.UTF-8
## tz Europe/Berlin
## date 2020-09-01
##
## ─ Packages ───────────────────────────────────────────────────────────────────
## package * version date lib source
## askpass 1.1 2019-01-13 [1] CRAN (R 4.0.0)
## assertthat 0.2.1 2019-03-21 [1] CRAN (R 4.0.0)
## backports 1.1.8 2020-06-17 [1] CRAN (R 4.0.0)
## bitops 1.0-6 2013-08-17 [1] CRAN (R 4.0.0)
## callr 3.4.3 2020-03-28 [1] CRAN (R 4.0.0)
## clamour * 0.1.0 2020-09-01 [1] Github (lazappi/clamour@c8ea1c7)
## cli 2.0.2 2020-02-28 [1] CRAN (R 4.0.0)
## colorspace 1.4-1 2019-03-18 [1] CRAN (R 4.0.0)
## crayon 1.3.4 2017-09-16 [1] CRAN (R 4.0.0)
## curl 4.3 2019-12-02 [1] CRAN (R 4.0.0)
## digest 0.6.25 2020-02-23 [1] CRAN (R 4.0.0)
## dplyr * 1.0.1 2020-07-31 [1] CRAN (R 4.0.2)
## ellipsis 0.3.1 2020-05-15 [1] CRAN (R 4.0.0)
## emo * 0.0.0.9000 2020-08-17 [1] Github (hadley/emo@3f03b11)
## evaluate 0.14 2019-05-28 [1] CRAN (R 4.0.0)
## fansi 0.4.1 2020-01-08 [1] CRAN (R 4.0.0)
## farver 2.0.3 2020-01-16 [1] CRAN (R 4.0.0)
## forcats * 0.5.0 2020-03-01 [1] CRAN (R 4.0.0)
## fs * 1.5.0 2020-07-31 [1] CRAN (R 4.0.2)
## generics 0.0.2 2018-11-29 [1] CRAN (R 4.0.0)
## ggforce 0.3.2 2020-06-23 [1] CRAN (R 4.0.2)
## ggplot2 * 3.3.2 2020-06-19 [1] CRAN (R 4.0.2)
## ggraph * 2.0.3 2020-05-20 [1] CRAN (R 4.0.0)
## ggrepel * 0.8.2 2020-03-08 [1] CRAN (R 4.0.0)
## ggtext * 0.1.0 2020-06-04 [1] CRAN (R 4.0.2)
## glue 1.4.1 2020-05-13 [1] CRAN (R 4.0.0)
## graphlayouts 0.7.0 2020-04-25 [1] CRAN (R 4.0.0)
## gridExtra 2.3 2017-09-09 [1] CRAN (R 4.0.0)
## gridtext 0.1.1 2020-02-24 [1] CRAN (R 4.0.2)
## gtable 0.3.0 2019-03-25 [1] CRAN (R 4.0.0)
## here * 0.1 2017-05-28 [1] CRAN (R 4.0.0)
## highr 0.8 2019-03-20 [1] CRAN (R 4.0.0)
## htmltools 0.5.0 2020-06-16 [1] CRAN (R 4.0.0)
## httr 1.4.2 2020-07-20 [1] CRAN (R 4.0.2)
## igraph * 1.2.5 2020-03-19 [1] CRAN (R 4.0.0)
## janeaustenr 0.1.5 2017-06-10 [1] CRAN (R 4.0.0)
## jsonlite 1.7.0 2020-06-25 [1] CRAN (R 4.0.0)
## kableExtra * 1.2.1 2020-08-27 [1] CRAN (R 4.0.2)
## knitr * 1.29 2020-06-23 [1] CRAN (R 4.0.0)
## labeling 0.3 2014-08-23 [1] CRAN (R 4.0.0)
## lattice 0.20-41 2020-04-02 [1] CRAN (R 4.0.0)
## lifecycle 0.2.0 2020-03-06 [1] CRAN (R 4.0.0)
## lubridate * 1.7.9 2020-06-08 [1] CRAN (R 4.0.0)
## magick * 2.4.0 2020-06-23 [1] CRAN (R 4.0.0)
## magrittr 1.5 2014-11-22 [1] CRAN (R 4.0.0)
## markdown 1.1 2019-08-07 [1] CRAN (R 4.0.0)
## MASS 7.3-51.6 2020-04-26 [1] CRAN (R 4.0.0)
## Matrix 1.2-18 2019-11-27 [1] CRAN (R 4.0.0)
## modeltools 0.2-23 2020-03-05 [1] CRAN (R 4.0.0)
## munsell 0.5.0 2018-06-12 [1] CRAN (R 4.0.0)
## NLP 0.2-0 2018-10-18 [1] CRAN (R 4.0.0)
## openssl 1.4.2 2020-06-27 [1] CRAN (R 4.0.0)
## pillar 1.4.6 2020-07-10 [1] CRAN (R 4.0.2)
## pkgconfig 2.0.3 2019-09-22 [1] CRAN (R 4.0.0)
## plyr 1.8.6 2020-03-03 [1] CRAN (R 4.0.0)
## png 0.1-7 2013-12-03 [1] CRAN (R 4.0.0)
## polyclip 1.10-0 2019-03-14 [1] CRAN (R 4.0.0)
## processx 3.4.3 2020-07-05 [1] CRAN (R 4.0.2)
## ps 1.3.3 2020-05-08 [1] CRAN (R 4.0.0)
## purrr * 0.3.4 2020-04-17 [1] CRAN (R 4.0.0)
## R6 2.4.1 2019-11-12 [1] CRAN (R 4.0.0)
## RColorBrewer * 1.1-2 2014-12-07 [1] CRAN (R 4.0.0)
## Rcpp 1.0.5 2020-07-06 [1] CRAN (R 4.0.0)
## RCurl 1.98-1.2 2020-04-18 [1] CRAN (R 4.0.0)
## reshape2 1.4.4 2020-04-09 [1] CRAN (R 4.0.0)
## rlang 0.4.7 2020-07-09 [1] CRAN (R 4.0.2)
## rmarkdown 2.3 2020-06-18 [1] CRAN (R 4.0.0)
## rprojroot 1.3-2 2018-01-03 [1] CRAN (R 4.0.0)
## rstudioapi 0.11 2020-02-07 [1] CRAN (R 4.0.0)
## rtweet * 0.7.0 2020-01-08 [1] CRAN (R 4.0.0)
## rvest * 0.3.6 2020-07-25 [1] CRAN (R 4.0.2)
## scales 1.1.1 2020-05-11 [1] CRAN (R 4.0.0)
## selectr 0.4-2 2019-11-20 [1] CRAN (R 4.0.0)
## sessioninfo 1.1.1 2018-11-05 [1] CRAN (R 4.0.0)
## slam 0.1-47 2019-12-21 [1] CRAN (R 4.0.0)
## SnowballC 0.7.0 2020-04-01 [1] CRAN (R 4.0.0)
## stringi 1.4.6 2020-02-17 [1] CRAN (R 4.0.0)
## stringr * 1.4.0 2019-02-10 [1] CRAN (R 4.0.0)
## tibble 3.0.3 2020-07-10 [1] CRAN (R 4.0.2)
## tidygraph 1.2.0 2020-05-12 [1] CRAN (R 4.0.0)
## tidyr * 1.1.1 2020-07-31 [1] CRAN (R 4.0.2)
## tidyselect 1.1.0 2020-05-11 [1] CRAN (R 4.0.0)
## tidytext * 0.2.5 2020-07-11 [1] CRAN (R 4.0.2)
## tm 0.7-7 2019-12-12 [1] CRAN (R 4.0.0)
## tokenizers 0.2.1 2018-03-29 [1] CRAN (R 4.0.0)
## topicmodels * 0.2-11 2020-04-19 [1] CRAN (R 4.0.0)
## tweenr 1.0.1 2018-12-14 [1] CRAN (R 4.0.0)
## usethis 1.6.1 2020-04-29 [1] CRAN (R 4.0.0)
## utf8 1.1.4 2018-05-24 [1] CRAN (R 4.0.0)
## vctrs 0.3.2 2020-07-15 [1] CRAN (R 4.0.2)
## viridis * 0.5.1 2018-03-29 [1] CRAN (R 4.0.0)
## viridisLite * 0.3.0 2018-02-01 [1] CRAN (R 4.0.0)
## webshot * 0.5.2 2019-11-22 [1] CRAN (R 4.0.0)
## withr 2.2.0 2020-04-20 [1] CRAN (R 4.0.0)
## wordcloud * 2.6 2018-08-24 [1] CRAN (R 4.0.0)
## xfun 0.16 2020-07-24 [1] CRAN (R 4.0.2)
## xml2 * 1.3.2 2020-04-23 [1] CRAN (R 4.0.0)
## yaml 2.2.1 2020-02-01 [1] CRAN (R 4.0.0)
##
## [1] /Library/Frameworks/R.framework/Versions/4.0/Resources/library